سندرم رابرت
سندرم رابرت که سندرم pseudothalidomide نیز نامیده می شود، یک اختلال ژنتیکی اتوزومال مغلوب نادر است که با عقب ماندگی خفیف تا شدید قبل از تولد یا اختلال در تقسیم سلولی مشخص می شود و منجر به ناهنجاری استخوان ها در جمجمه، صورت، بازوها و پاها می شود.
این بیماری در اثر جهش در ژن ESCO2 ایجاد می شود. این بیماری یکی از نادرترین اختلالات اتوزومال مغلوب است. جهش باعث می شود تقسیم سلولی به آهستگی یا ناهموار اتفاق بیفتد و سلول های دارای محتوای ژنتیکی غیر طبیعی بمیرند.
سندرم رابرت می تواند هم در مردان و هم در زنان تأثیر بگذارد. اگرچه این اختلال نادر است، اما گروه مبتلا متنوع هستند. میزان مرگ و میر در افراد به شدت آسیب دیده زیاد است. این سندرم پس از جراح و پزشک آمریکایی جان بینگام رابرتز (1924-1962) نامگذاری شده است که اولین بار در سال 1919 آن را توصیف کرد.
در زیر لیستی از علائم مرتبط با سندرم رابرت آورده شده است:
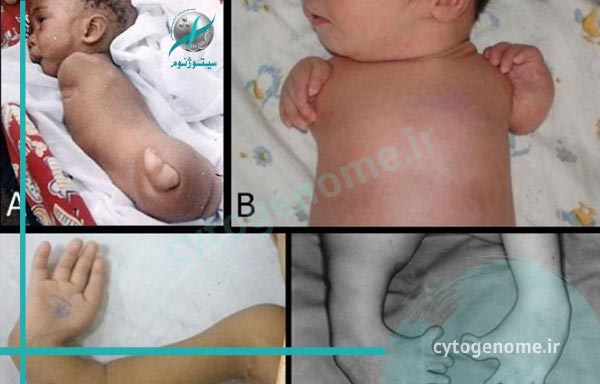
- tetraphocomelia متقارن دو طرفه – نقص هنگام تولد که در آن دست و پا به بازوها و پاهای کوتاه شده وصل می شوند
- عقب ماندگی رشد قبل از تولد
- هیپوپلازی – توسعه ناقص یک بافت یا اندام؛ کمتر از شدت آپلازی، که به هیچ وجه توسعه ای ندارد
- الیگوداستیلی – تعداد انگشتان دست یا انگشتان پا کمتر است
- آپلازی انگشت شست – عدم وجود انگشت شست
- Syndactyly- شرایطی که دو یا چند انگشت (یا انگشتان پا) به یکدیگر وصل می شوند. اتصال ممکن است استخوان ها یا فقط پوست بین انگشتان را درگیر کند
- كلينوداكستي- خميدگي انگشت پنجم (انگشت كوچك) به سمت انگشت چهارم (انگشت حلقه) به علت عدم توسعه استخوان مياني در انگشت پنجم
- انقباضات خم شدن آرنج / زانو – عدم توانایی در صاف کردن کامل بازو یا پا
- شکاف لب – وجود یک یا دو شکاف عمودی در لب فوقانی؛ می تواند از یک طرف باشد (یک طرفه) یا هر دو طرف (دو طرفه)
- شکاف کام – باز در سقف دهان
- برآمدگی قبل از عمل – قسمت فوقانی دهان دورتر از قسمت تحتانی دهان است
- میکروگناتیا – چانه کوچک
- میکروبراسیفالی – از اندازه سر معمولی کوچکتر است
- هیپوپلازی مالار – توسعه نیافتگی استخوان های گونه
- شکستگی شکاف پایین دست – گوشه های بیرونی چشم ها به سمت پایین نشان می دهد
- پرفشاری خون چشمی – چشمی غیرمعمول گسترده
- Exophthalmos – یک چشم بزرگ بیرون زده
- بستن قرنیه – بستن قسمت جلویی چشم
- باریک شدن بینی – بینی هیپوپلاستیک باعث کاهش عرض بینی می شود
- بینی منقار – بینی با یک پل برجسته که ظاهر منحنی آن را نشان می دهد
- ناهنجاری های گوش
- ناتوانی ذهنی
- آنسفالوسل (فقط در موارد شدید) – نقص نادر لوله عصبی که با بیرون زدگی های کیسه مانند مغز مشخص می شود
- مرگ و میر در میان کسانی که به شدت تحت تأثیر سندرم رابرتز قرار دارند زیاد است. با این حال، افراد خفیف مبتلا ممکن است تا بزرگسالی زنده بمانند
وراثت در سندرم رابرت
ESCO2 ، واقع در کروموزوم انسانی 8، به عنوان ژن مسئول سندرم رابرت شناخته شده است. در حقیقت، ESCO2 تنها ژن شناخته شده ای است که جهش های ایجاد کننده RBS را نشان داده است. همچنین، تمام افرادی که از نظر سیتوژنتیک به سندرم رابرتز مبتلا شده اند نیز در ژن ESCO2 جهش داشته اند.
برای بروز سندرم رابرتز، كودك باید ژن معیوب را به روش اتوزومال مغلوب به ارث ببرد. به عبارت دیگر، کودک باید دو نسخه از ژن معیوب (یکی از هر والدین) به ارث ببرد. ژن ESCO2 در تقسیم سلولی در بیماران مبتلا به سندرم رابرتز تأثیر ویژه ای دارد. در تقسیم سلولی معمولی، هر کروموزوم کپی می شود و سپس به کپی ریز تشکیل شده در سانترومر (قسمت مرکزی یک کروموزوم) وصل می شود. با این حال، در تقسیم سلولی سندروم رابرت، نسخه ها معمولاً در مركز متصل نمی شوند. در نتیجه، کروموزوم ها به درستی صاف نمی شوند و همین امر باعث می شود سلول بسیار آهسته تقسیم شود یا حتی به هیچ وجه تقسیم نشود. سلول های جدید به طور معمول کروموزوم های خیلی کمی دارند. تعداد عجیب کروموزوم ها باعث از بین رفتن سلول های معیوب می شود که منجر به ناهنجاری های مرتبط با سندرم رابرتز می شود.
بسیاری از ناهنجاری های جسمی مرتبط با سندرم رابرت بسیار شبیه به ناهنجاری هایی است که در کودکانی رخ می دهد که مادران آن ها در دوران بارداری از تالیدومید استفاده می کردند. شباهت های بدنی نشان می دهد که یک زیست شناسی اساسی مشابه بین ESCO2 و تالیدومید وجود دارد. در نتیجه فرض بر این است که تالیدومید بر روی کروموزوم ها و تقسیم سلولی به روشی مشابه با ESCO2 تأثیر می گذارد. به همین دلیل، گاهی به سندرم رابرت سندرم سودومیدالیدید گفته می شود.
کشف سندرم رابرت
کشف ESCO2 به عنوان ژن مسئول سندرم رابرت با مطالعه نمونه هایی از پانزده خانواده تحت تأثیر سندرم رابرتز انجام شد. در سال 1995، هوگو وگا و میریام گوردیلو، دو متخصص ژنتیک کلمبیایی، تصمیم گرفتند سندرم رابرتز را کاملاً درک کنند. وگا و گوردیلو تعداد بسیار زیادی از بیماران مبتلا به سندرم رابرت را در دانشگاه Universidad Nacional de Colombia مشاهده کردند. این دو متخصص ژنتیک کلمبیایی در مجموع هفت خانواده مبتلا به سندرم رابرتز را دقیقاً در خارج از بوگوتا پیگیری کردند و فهمیدند که چهار فرد از این هفت خانواده یک جد مشترک مشترک در قرن 18 دارند. با استفاده از این اطلاعات، وگا و گوردیلو توانستند ژن مسئول سندرم رابرت را که ESCO2 بود، تشخیص دهند.
تشخیص بالینی
تشخیص بالینی سندرم رابرتز در افرادی که دارای عقب ماندگی مشخصه رشد قبل از تولد، ناهنجاری های اندام و ناهنجاری های استخوان ران است انجام می شود. خصوصیات خاصی که در تشخیص بالینی جستجو می شود که در زیر آورده شده است.
- عقب ماندگی رشد قبل از تولد – طول و وزن کم هنگام تولد که می تواند از خفیف تا شدید باشد
- ناهنجاری های اندام – تترافوکوملیای متقارن دو طرفه ، الیگوداستیلی ، آپلازی انگشت شست ، سینداستیلی ، کلینوداستیلی و انقباضات خم آرنج و زانو
- ناهنجاری های کرانیوفیزیال – شکاف لب و کام دو طرفه ، میکروگنیاتیا ، فشار خون بالا ، اگزوفتالموس ، شکاف های پالپ مغزی پایین ، هیپوپلازی ملار ، هیپوپلازی بینی ، و علائم گوش
تشخیص قطعی سندرم رابرتز به آزمایش سیتوژنتیک خون محیطی متکی است.
آزمایش سیتوژنتیک
آماده سازی سیتوژنتیک که توسط تکنیک Giemsa یا C باند رنگ آمیزی شده است، دو ناهنجاری کروموزومی مشخصه را نشان می دهد. اولین ناهنجاری کروموزومی، جداسازی زودرس سانترومر (PCS) نامیده می شود و محتمل ترین مکانیسم بیماری زا برای سندروم رابرتز است. کروموزوم هایی که دارای PCS هستند، به جای آنافاز (یک فاز زودتر از کروموزوم های معمولی)، سانترومرهای خود را در طول متافاز جدا می کنند. دومین اختلال کروموزومی، دافعه هتروکروماتین (HR) نام دارد. کروموزوم هایی که دارای HR هستند در طول متافاز جداسازی مناطق هتروکرومیک را تجربه می کنند. کروموزوم هایی که دارای این دو ناهنجاری هستند، به دلیل عدم وجود تنگی و دفع اولیه در مناطق هتروکروماتیک ظاهر “مسیر ریلی” را نشان می دهند. نواحی هتروکرومی مناطقی در نزدیکی سانترومرها و سازمان دهنده های هسته است. وضعیت حامل را نمی توان با آزمایش سیتوژنتیک تعیین کرد. سایر یافته های رایج آزمایش سیتوژنتیک در بیماران مبتلا به سندرم رابرتز در زیر آورده شده است.
- Aneuploidy- بروز یک یا چند کروموزوم اضافی یا مفقود شده
- ریز هسته – هسته کوچکتر از حد طبیعی است
- هسته چند هسته ای – هسته دارای بیش از یک لوب است
آزمایش ژنتیک
در این زمان، ESCO2 تنها ژن شناخته شده ای است که باعث جهش سندرم رابرتز می شود. همچنین، تمام افرادی که با تکنیک سیتوژنتیک به سندرم رابرتز مبتلا شده اند نیز دارای جهش ESCO2 هستند. تأیید تشخیص سندرم رابرت مستلزم تشخیص ناهنجاری های کروموزومی مشخصه (PCS و HR) یا شناسایی دو جهش ESCO2 است که با سندرم رابرتز در ارتباط بوده اند.
آزمایش حامل و تشخیص بارداری
آزمایش حامل سندرم رابرت نیاز به شناسایی قبلی جهش ایجاد کننده بیماری در خانواده دارد. ناقلین این اختلال به دلیل ماهیت مغلوب اتوزومال بیماری، هتروزیگوت هستند. حاملان همچنین در معرض خطر ابتلا به خود سندرم رابرتز نیستند. تشخیص پیش از تولد سندرم رابرتز نیاز به معاینه سونوگرافی دارد که همراه با آزمایش سیتوژنتیک یا شناسایی قبلی جهش های ESCO2 ایجاد کننده بیماری در خانواده انجام می شود.
اختلالات مرتبط با ژنتیک
در این زمان، هیچ فنوتیپ دیگری (بیان قابل مشاهده از یک ژن) وجود ندارد که برای جهش در ژن ESCO2 کشف شده باشد.
تشخیص های افتراقی سندرم رابرت
در موارد ناهنجاری خفیف، اختلالات زیر باید در تشخیص افتراقی در نظر گرفته شود:
- سندرم Baller-Gerold
- آنمی فانکونی (FA)
در موارد تظاهرات شدید، در تشخیص افتراقی باید اختلالات زیر را در نظر گرفت:
- سندرم تشعشع ترومبوسیتوپنی-پرتونگار
- Tetra-Amelia ، X- مرتبط
- Tetra-Amelia ، اتوزومال مغلوب
- اسپلوژنوگنادال فیوژن با نقص اندام و میکروگناتیا
- سندرم DK Phocomelia
- سندرم هولت-اورام
- جنینوپاتی تالیدومید
در موارد مشابه یافته های سیتوژنتیک، اختلالات زیر را باید در تشخیص افتراقی در نظر گرفت:
- سندرم کرنلیه د لانژ (CdLS)
- سندرم آنیوپلوئیدی واریزی شده موزاییک
شیوع
سندرم رابرت یک بیماری بسیار نادر است که فقط در حدود 150 فرد گزارش شده است. اگرچه فقط حدود 150 مورد گزارش شده است، اما گروه مبتلا کاملاً متنوع است و در سرتاسر جهان رواج دارد. رابطه فامیلی والدین (والدین با هم ارتباط نزدیکی دارند) با این اختلال ژنتیکی در ارتباط است.

