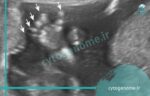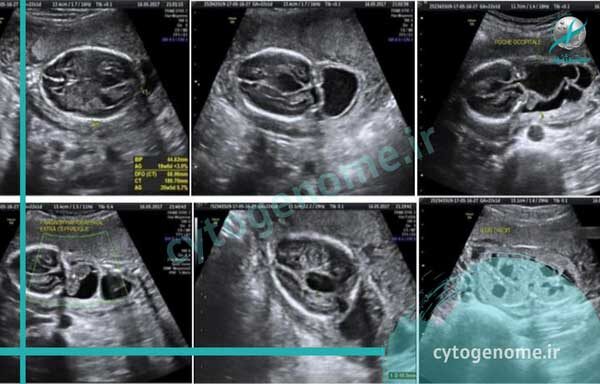
سندروم مکل گروبر (MKS) یک بیماری کشنده، نادر ژنتیکی است که با الگوی توارث اتوزومال مغلوب منتقل می شود و با سه علامت رایج یعنی: انسفالوسل اکسیپیتال، کلیه های پلی کیستیک بزرگ و پلی داکتیلی پس محوری شناسایی می شود. مرگ مبتلایان معمولاً در رحم (در دوران جنینی) یا اندکی پس از تولد اتفاق می افتد و کم پیش می آید که مبتلایان وارد سنین بالاتر شوند. ژنتیک دانان علت سندروم مکل را جهش های ژنتیکی در حدود 13 ژن می دانند. با این مقاله از آزمایشگاه ژنتیک سیتوژنوم همراه باشید تا اطلاعات بیشتری در مورد علل و فراوانی سندروم مکل کسب کنید.
چقدر احتمال دارد فرزندان من به سندروم مکل مبتلا شوند؟
سندروم مکل (Meckel syndrome) مردان و زنان را به تعداد مساوی مبتلا می کند و از هر 13250 یا از هر 140000 نفر 1 نفر را در سراسر جهان درگیر کرده و بروز آن در جمعیت های خاصی شایع تر است. به عنوان مثال، این عارضه حدود 1 نفر از هر 9000 نفر فنلاندی و حدود 1 نفر از هر 3000 نفر بلژیکی تبار را تحت تاثیر قرار می دهد. نکته مهم در مورد این اختلال این است که علت سندروم مکل جهش ژنتیکی است و این جهش توارث اتوزوم مغلوب دارد؛ بنابراین در افرادی که ازدواج فامیلی داشته اند و سابقه این بیماری را داشته اند رایج تر است. اختلالات ژنتیکی مغلوب زمانی اتفاق می افتند که یک فرد، ژن جهش (تغییر) یافته یکسانی را برای یک صفت از هر یک از والدین به ارث ببرد. اگر فردی یک ژن طبیعی و یک ژن تغییر یافته برای بیماری دریافت کند، ناقل بیماری خواهد بود اما علائمی از خود نشان نخواهد داد. خطر این که دو والدین ناقل، هر دو ژن تغییر یافته را به فرزندشان منتقل کنند و در نتیجه فرزندی مبتلا داشته باشند در هر بارداری 25 درصد است. خطر داشتن فرزندی که مانند والدین ناقلِ بدون علامت است در هر بارداری 50 درصد است. در نهایت شانس دریافت ژن های طبیعی از هر دو والدین برای کودک 25 درصد خواهد بود.
علل سندروم مکل چیست؟
علت سندروم مکل جهش ژنتیکی در یک یا چند مورد از 13 ژنی است که تا به حال شناسایی شده اند. این ژن ها عبارت هستند از:
- B9D1
- B9D2
- CC2D2A
- CEP290
- MKS1 (رایج ترین ژن)
- RPGRIP1L
- TCTN2
- TCTN3
- TMEM67
- TMEM107
- TMEM216
- TMEM2
جهش در این 13 ژن، 75 درصد از کل موارد ابتلا به سندروم مکل- گروبرت را تشکیل می دهد. 25 درصد باقی مانده نیز دلایل و ریشه ژنتیکی ناشناخته دارند. بسیاری از این ژن ها همچنین مسئول ایجاد یک بیماری عصبی به نام سندرم ژوبرت (Joubert syndrome) هستند که منجر به این مفهوم می شود: سندرم مکل شکل شدید مرگبار سندرم ژوبرت است. پروتئین های تولید شده توسط این ژن ها بر ساختار یا عملکرد سلولی به نام “مژک های اولیه” یا سیلیای اولیه (Primary cilia) تأثیر می گذارند. مژک ها برآمدگی های میکروسکوپی هستند که از روی سطح سلول بیرون می آیند و به انتقال اطلاعات در مسیرهای سیگنال دهی کمک می کنند. مژک (یا سیلیا) برای بسیاری از عملکردهای سلولی، در بسیاری از انواع سلول ها به ویژه سلول های کلیوی، کبدی، چشم و مغز مهم است. جهش در این ژن ها باعث ایجاد مشکلاتی در عملکرد مژک های اولیه می شود و در نتیجه نقایص مختلفی بسته به نوع سلول درگیر، ایجاد می شود. عملکرد معیوب مژک های اولیه می تواند مسئول ناهنجاری های رشدی به ویژه در کلیه ها، مغز و قلب باشد.
اختلالات مرتبط
در بالا توضیح دادیم که علائم و علت سندروم مکل چیست، اما باید بدانید که هر گردی گردو نیست و هرگاه که علائم بالا دیده شد، به این معنا نیست که فرد به مکل مبتلاست. گروهی از اختلالات ژنتیکی وجود دارند که علائمشان می تواند مشابه علائم سندرم مکل باشد. مقایسه این علائم و بررسی ژنتیکی می تواند برای تشخیص افتراقی بین مکل و دیگر اختلالات مفید باشد. بعضی از بیماری هایی که می توانند علائم شبیه سندرم مکل داشته باشند عبارت هستند از:
- سندروم اسمیت لملی اوپیتز (SLOS): یک اختلال رشدی، ارثی و نادر می باشد که مشخصه آن وجود ناهنجاری های متعددی از بدو تولد (مادرزادی) است. علائم این سندروم ممکن است شامل وجود ویژگی های مشخص در صورت، میکروسفالی، محدودیت رشد، ناتوانی ذهنی، داشتن انگشت اضافه در دست یا پا (پلی داکتیلی)، از دست دادن بینایی، رشد ناقص اندام تناسلی مردانه، بینی کوتاه با سوراخ های بینی جابجا شده و تنگی دریچه پیلور (دهانه معده) باشد. در برخی از بیماران، ناهنجاری های مغزی یا قلبی ممکن است وجود داشته باشد؛ به هر حال علائم خاص ناشی از این بیماری در هر فرد بسیار متفاوت است. سندرم اسمیت لملی اپتیز نیز با الگوی اتوزومال مغلوب به ارث می رسد.
- سندروم پاتائو: سندروم پاتائو یا تریزومی 13 یک اختلال کروموزومی است که طی آن به جای داشتن 2 نسخه از کروموزوم 13، در هر سلول 3 نسخه (تریزومی) وجود دارد. در برخی از افراد مبتلا، تنها درصدی از سلول ها ممکن است حاوی کروموزوم 13 اضافی (موزاییکیسم) باشند، و سلول های دیگر حاوی همان 2 کروموزومی طبیعی هستند. در بسیاری از نوزادان و کودکان مبتلا به تریزومی 13، ناهنجاری های ممکن است شامل تأخیر رشد، ناتوانی ذهنی شدید، چشم های کوچک غیرعادی (میکروفتالمی)، شیار غیرطبیعی در لب بالایی (شکاف لب)، بسته شدن ناقص سقف دهان (شکاف کام)، بیضه های نزول نکرده (کریپتورکیدیسم) در مردان مبتلا، وجود انگشتان اضافه در دست و پا اضافی (پلی داکتیلی) باشد. ناهنجاری هایی دیگری نیز ممکن است در ناحیه سر و صورت وجود داشته باشد مانند: سر نسبتاً کوچک (میکروسفالی) با پیشانی شیب دار، بینی پهن و صاف، چشم های دور از هم (هیپرتلوریسم چشمی)، چین های عمودی پوست که چشم ها را می پوشاند (چین های اپیکانتال)، نقص پوست سر و گوش های ناقص و کم تنظیم. نوزادان مبتلا همچنین ممکن است رشد ناقص مناطق خاصی از مغز (مانند مغز جلویی)، ناهنجاری های کلیوی و نقص ساختمانی قلبی از بدو تولد (مادرزادی) داشته باشند. عوارض تهدید کننده زندگی ممکن است در دوران نوزادی یا اوایل کودکی ایجاد شوند.
- سندروم های دنده کوتاه: یا پلی داکتیلی؛ گروهی از اختلالات اسکلتی نادر هستند که با کمبود رشد و در نتیجه کوتاهی قد، قفسه سینه باریک، دنده های کوتاه غیر طبیعی و انگشتان اضافه در دست و پا (پلی داکتیلی) مشخص می شوند. همپوشانی قابل توجهی در علائم مرتبط این سندروم با سندروم مکل وجود دارد که از جمله آن ها می توان به: کلیه های پلی کیستیک، توسعه نیافتگی (هیپوپلازی) ریه ها، ناهنجاری های ادراری تناسلی، ناهنجاری های سیستم عصبی مرکزی، تاخیر در رشد و شکاف لب و شکاف کام اشاره کرد.
نویسنده و مترجم: دکتر سارا فرخی مشاور ژنتیک