پاراپلژی اسپاستیک ارثی (Hereditary spastic paraplegia که اختصارا HSP گفته میشود) را با نامهایی همچون پاراپارزی اسپاستیک خانوادگی (FSP)، سندروم استرومپل لورن (Strümpell-Lorrain syndrome)، فلج ناقص و یا اسپاسم وراثتی میشناسند. این بیماری یک اختلال ارثی نادر است که باعث ضعف و سفتی عضلات پا میشود و علائم آن به تدریج در طول زمان بدتر میگردد. تشخیص دقیق افراد مبتلا به بیماری HSP بدون انجام تست ژنتیک دشوار است زیرا اغلب این بیماری را با بیماریهای دیگر اشتباه میگیرند. با این مقاله از مجله پزشکی آزمایشگاه ژنتیک پزشکی سیتوژنوم همراه باشید تا بیشتر با علائم و علت پاراپلژی اسپاستیک ارثی آشنا شوید.
بیماری پاراپلژی اسپاستیک ارثی یا سندروم استرومپل لورن چیست و انواع آن کدامند؟
پاراپلژی اسپاستیک ارثی (HSP) به گروهی از اختلالات ارثی اشاره دارد که با علامتی مثل ضعف پیشرونده و اسپاستیسیته (سفتی) پاها مشخص میشود. در اوایل دوره بیماری، ممکن است مشکلات خفیف راه رفتن و سفتی در پاها احساس شود اما این علائم معمولاً به آرامی پیشرفت میکنند به طوری که در نهایت افراد مبتلا به HSP ممکن است برای راه رفتن به عصا، واکر یا ویلچر نیاز داشته باشند. اگرچه ویژگیهای اولیه بیماری HSP، اسپاستیسیته و ضعف پیشرونده اندامهای تحتانی است، اما اگر فرد به اَشکال پیچیده این اختلال مبتلا باشد ممکن است علائم دیگری را نیز تجربه کند. این علائم اضافی شامل اختلال بینایی به دلیل آب مروارید و مشکلات عصب بینایی و شبکیه چشم، آتاکسی (عدم هماهنگی عضلات)، صرع، اختلال شناختی، نوروپاتی محیطی و ناشنوایی است. لازم است بدانید که این بیماری به 2 دسته تقسیم میشود:
- نوع خالص یا ساده
- نوع پیچیده
علائم پاراپلژی اسپاستیک خانوادگی
همانطور که گفته شد، پاراپلژی اسپاستیک ارثی دو نوع دارد که در ادامه علائم آنها را میخوانیم:
الف) علائم فلج ناقص خالص: حدود 90 درصد از افراد مبتلا به سندروم استرومپل لورن دارای نوع خالص این بیماری هستند. در HSP خالص علائم به طور کلی محدود به ضعیف شدن تدریجی در پاها، اختلال مثانه، اسپاستیسیته، راه رفتن غیر طبیعی، کاهش حس تعادل و گاهی اوقات اختلال در احساسات پاها میباشد. اگر بخواهیم این علائم را دسته بندی کنیم، به صورت زیر خواهد بود:
- ضعف تدریجی در پاها
- افزایش تون عضلانی
- سفتی عضلات (اسپاستیسیته)
- مشکلاتی در ادرار کردن مانند نیاز فوری به ادرار کردن حتی زمانی که مثانه پر نیست
- کاهش حس در پاها (گاهی اوقات)
- کودکان ممکن است دچار سفتی پا و مشکلاتی در راه رفتن شوند، مانند زمین خوردن به ویژه در زمینهای ناهموار. این اتفاق بدان دلیل است که خم کردن انگشتان پا به سمت بالا به دلیل داشتن عضلات ضعیف برای آنها دشوار است.
- برخی از افراد ممکن است در نهایت نیاز به استفاده از عصای پیاده روی یا ویلچر داشته باشند. هرچند برخی دیگر ممکن است نیازی به استفاده از هیچ نوع تجهیزات کمک حرکتی نداشته باشند
ب) علائم فلج ناقص پیچیده: 10 درصد باقیمانده مبتلایان اسپاسم وراثتی به شکل پیچیده این بیماری دچار هستند. در HSP پیچیده که یک اختلال نادرتر است، ممکن است علائم بیشتری نسبت به نوع خالص وجود داشته باشد که شامل موارد زیر میباشد:
- نوروپاتی محیطی (آسیب عصبی در پاها یا سایر اندامها)
- صرع
- آتاکسی (مشکلاتی در ایجاد تعادل، هماهنگی و گفتار)
- نوروپاتی بینایی (آسیب به عصب بینایی)
- رتینوپاتی (آسیب به شبکیه)
- زوال عقل
- ایکتیوز (وضعیتی که باعث خشک و ضخیم شدن گسترده و مداوم پوست میشود)
- عقب ماندگی ذهنی
- ناشنوایی
- مشکلاتی در گفتار، بلع یا تنفس
مبتلایان به نوع پیچیده ممکن است طیف وسیعی از علائم ذکر شده را (از خفیف تا شدید) از خود بروز بدهند.
اگر به دنبال بررسی راههای درمان این بیماری هستید روی لینک زیر کلیک کنید:
سن شروع سندروم استرومپل لورن
در گذشته گفته میشد که سن شروع سندروم استرومپل لورن در اوایل دوران کودکی یا در برخی موارد شروع دیرتر در بزرگسالی میباشد. یعنی سن شروع دو نقطه حداقل و حداکثر دارد که بین بازههای 2 سالگی و حدود 40 سالگی است. اما امروزه یافتههای جدید نشان میدهند که شروع زودتر منجر به تاخیر در بروز علائم بیماری بدون از دست دادن تعادل یا نیاز به استفاده از ویلچر میشود و افرادی که در سنین بالاتر علائم را نشان می دهند، علائمشان با سرعت بیشتری وخیم میشود.
علت پاراپلژی اسپاستیک ارثی چیست؟
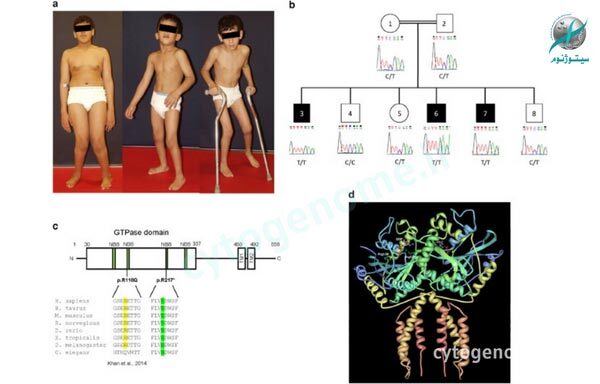
همانطور که از نام این بیماری میتوان حدس زد، علت پاراپلژی اسپاستیک ارثی “جهش ژنتیکی” در یک یا چند ژن مرتبط میباشد؛ بنابراین این بیماری از قوانین اصلی وراثت پیروی میکند و میتواند به صورت اتوزومال غالب، اتوزومال مغلوب یا مغلوب وابسته به X به ارث برسد. لازم به ذکر است که نحوه وراثت تأثیر مستقیمی بر شانس ارث بردن این اختلال دارد. به طور کلی اکثر افراد مبتلا به HSP خالص، یک ژن معیوب را از یکی از والدین خود به ارث بردهاند (توارث اتوزوم غالب)، اما افراد مبتلا به شکل پیچیده این بیماری معمولاً دو ژن معیوب در هر سلول بدن خود دارند که هر یک از این 2 ژن را از دو والدشان به ارث گرفتهاند (یعنی یک ژن از پدر و یک ژن از مادر که به معنای توارث اتوزوم مغلوب است). این جهشهای ژنی باعث میشوند ناهنجاریهای موجود در اعصاب طولانی در ستون فقرات بدتر شود؛ این اعصاب به طور معمول تون عضلات و حرکت در قسمت پایین تنه را کنترل میکنند.
در حال حاضر بیش از 70 ژنوتیپ برای این اختلال توصیف شده است و بیش از 50 جایگاه ژنتیکی برایش مشخص گشته. 10 ژن با توارث اتوزومال غالب شناسایی شده است که یکی از این موارد ژن SPG4 میباشد که حدود 50 درصد از کل موارد ابتلا با این الگو (یعنی اتوزوم غالب) و تقریباً 25 درصد از کل موارد HSP را تشکیل میدهد. 12 ژن نیز شناسایی شدهاند که به صورت اتوزومی مغلوب به ارث میرسند؛ این گروه حدود یک سوم موارد ابتلا را تشکیل میدهند.
همه گیرشناسی پاراپلژی اسپاستیک ارثی
در سرتاسر جهان، شیوع همه پاراپلژی های اسپاستیک ارثی در مجموع 2 الی 6 مورد در هر 100000 نفر تخمین زده میشود. یک مطالعه نروژی بر روی بیش از 2.5 میلیون نفر که در مارس 2009 منتشر شد، نرخ شیوع HSP را 7.4 در 100000 نفر (صد هزار نفر) از جمعیت نشان داد که نرخی بالاتر اما در محدوده مشابه مطالعات قبلی بود. هیچ تفاوتی در میزان بروز پاراپلژی اسپاستیک ارثی مربوط به جنسیت یافت نشد (زن و مرد به یک میزان در معرض خطر ابتلا هستند) و میانگین سن شروع علائم 24 سال اعلام شد.
جمع بندی
پاراپارزی اسپاستیک خانوادگی یا سندروم استرومپل لورن گروهی از بیماریهای ارثی هستند که ویژگی اصلی آنها اختلال پیشرونده راه رفتن است. این بیماری با سفتی پیشرونده (اسپاستیسیته) و انقباض در اندام تحتانی خود را نشان میدهد. تشخیص HSP در درجه اول با معاینه عصبی و آزمایش برای رد سایر اختلالات مشابه است؛ دیدن ناهنجاریهایی در MRI مغز مانند جسم پینهای نازک، ممکن است در برخی از اشکال پیچیده HSP دیده شود. چندین جهش ژنتیکی شناسایی شده است که زمینه ساز اشکال مختلف HSP است و آزمایش و تشخیص ژنتیکی تخصصی در برخی از مراکز پزشکی برای تایید نهایی موجود است. HSP انواع مختلفی از وراثت دارد و همه کودکان یک خانواده لزوماً علائمی ندارند، اگرچه ممکن است ناقل ژن غیر طبیعی باشند (توارث مغلوب). بسته به ژن خاصی که درگیر شده، علائم فلج ناقص ممکن است در دوران کودکی یا بزرگسالی شروع شود. شما عزیزان در صورتیکه به اطلاعات بیشتری در مورد این بیماری نیاز دارید میتوانید با مشاور ژنتیک ما در آزمایشگاه سیتوژنوم بصورت تلفنی در ارتباط باشید.
نویسنده و مترجم: دکتر سارا فرخی مشاور ژنتیک