آتاکسی به معنای فقدان هماهنگی بین عضلات ارادی (مخطط) و از دست دادن کنترل حرکت است که بر ثبات راه رفتن، حرکت چشم ها و گفتار افراد تأثیر می گذارد. آتاکسی مغزی نخاعی یا همان نخاعی مخچه ای (SCA) یک بیماری ژنتیکی و ارثی، پیش رونده، تخریب کننده عصبی و هتروژن است که عمدتاً مخچه را درگیر می کند. SCA زیر مجموعه ای از آتاکسی های ارثی مخچه می باشد و یک بیماری نادر به حساب می آید. تا به امروز، بیش از 40 نوع ژنتیکی از SCA متمایز شناسایی شده است که بر اساس جایگاه های ژنتیکی به ترتیب شناساییشان طبقه بندی می شوند. آزمایشگاه ژنتیک پزشکی سیتوژنوم در این مقاله به بررسی آسیب شناسی این بیماری خواهد پرداخت.
آتاکسی مغزی نخاعی چیست؟
آتاکسی مغزی نخاعی یا آتاکسی های نخاعی مخچه ای (SCAs) یک گروه ژنتیکی ناهمگن از اختلالات پیشرونده ارثی هستند که مشخصه بالینی آن ها از دست دادن تعادل و هماهنگی همراه با گفتار نامفهوم است و شروع علائم آن اغلب در بزرگسالی است. ویژگی دیگر این بیماری دژنراتیو بودن آن است، یعنی موجب تحلیل رفتن بافت آسیب دیده و عدم کارکرد طبیعی آن می شود. آتاکسی نخاعی مخچه ای چندین نوع مختلف دارد و محققان تا به الان 40 نوع از آن ها را شناسایی و نام گذاری کرده اند و هر کدام می توانند به تنهایی یک بیماری عصبی جداگانه در نظر گرفته شوند. تخمین زده می شود که 150000 نفر در ایالات متحده به آتاکسی نخاعی مخچه ای مبتلا هستند. این بیماری شدید و غالبا کشنده است و هیچ درمان قطعی برای آن در دسترس نمی باشد. SCA می تواند هر فردی را در هر سنی تحت تاثیر قرار دهد و در بسیاری از موارد، افراد تا زمانی که فرزندان مبتلا که علائم ابتلا به این اختلال را نشان بدهد به دنیا نیاورند، از حامل و ناقل بودن خود آگاه نمی شوند.
علت آتاکسی مغزی نخاعی چیست؟
علت آتاکسی مغزی نخاعی همانطور که در سایر اشکال آتاکسی دیده می شود، آتروفی مخچه است که در اثر یک جهش ژنتیکی ایجاد می شود. شروع علائم آتاکسی مغزی نخاعی معمولاً بعد از سن 18 سالگی است و به کندی پیشرفت می کند و علائم در طی چند سال بدتر می شوند. لازم به ذکر است که برخی از انواع SCA می توانند با سرعت بیشتری پیشرفت کنند. به هر حال جهش در ژنی به نام ATXN1 باعث ایجاد SCA1 (آتاکسی مغزی نخاعی نوع 1) می شود؛ ژن ATXN1 دستورالعمل هایی را برای ساخت پروتئینی به نام آتاکسین-1 ارائه می دهد. این پروتئین در سراسر بدن یافت می شود، اما عملکرد آن ناشناخته است؛ خوب است بدانید که آتاکسین-1 در داخل هسته سلول ها قرار دارد. محققان بر این باورند که آتاکسین-1 ممکن است در تنظیم فعایت های مختلف منتهی به تولید پروتئین ها، از جمله مرحله اول تولید پروتئین (رونویسی) و پردازش RNA نقش داشته باشد.
جهش های ژن ATXN1 که باعث ایجاد SCA1 می شوند در ناحیه ای از DNA است که شامل تکرار سه نوکلئوتیدی CAG می باشد. این بخش از یک کدون سه تایی (شامل سیتوزین، آدنین و گوانین) تشکیل شده است که چندین بار پشت سر هم تکرار می شوند. به طور معمول، توالی CAG در بدن یک فرد سالم بین 4 تا 39 بار در ژن تکرار می شود اما در افراد مبتلا به آتاکسی مغزی نخاعی نوع1 ، توالی CAG حدود 40 تا بیش از 80 بار تکرار شده است. افرادی که 40 الی 50 تکرار در بدن خود دارند علائم و نشانه های SCA1 را در اواسط بزرگسالی تجربه می کنند، در حالی که افراد با بیش از 70 تکرار معمولاً علائم و نشانه ها را در دوران نوجوانی خود بروز خواهند داد. افزایش طول توالی CAG منجر به تولید یک نسخه طولانی و غیر طبیعی از پروتئین آتاکسین 1 می شود که در قالب سه بعدی اشتباهی تا می شود. این پروتئین غیرطبیعی با سایر پروتئین ها جمع می شود تا توده هایی (انباشتگی) در هسته سلول ها ایجاد کند. این توده ها از عملکرد طبیعی پروتئین آتاکسین-1 جلوگیری می کنند که به سلول ها آسیب می رساند و منجر به مرگ سلولی می شود. به دلایلی که هنوز مشخص نیست، تجمعات آتاکسین-1 فقط در مغز و نخاع (سیستم عصبی مرکزی) یافت می شود. سلول های مخچه (بخشی از مغز است که حرکات را هماهنگ می کند)، به طور خاص به تغییرات در شکل و عملکرد آتاکسین-1 حساس هستند. با گذشت زمان، از دست دادن سلول های مخچه باعث مشکلات حرکتی که مشخصه SCA1 است می شود.
علائم آتاکسی نخاعی مخچه ای چیست؟
علائم و نشانه های آتاکسی مغزی نخاعی معمولاً بعد از 18 سالگی ظاهر می شوند و به آرامی در طی چندین سال بدتر می شوند. علائم این بیماری اغلب شامل موارد زیر است:
- حرکات غیر ارادی چشم
- هماهنگی ضعیف بین دست و چشم
- وجود مشکلاتی بین تعادل و هماهنگی اعضای بدن
- لکنت زبان
- مشکل در پردازش و به خاطر سپردن اطلاعات (اختلالات یادگیری).
- راه رفتن ناهماهنگ، نا موزون و نا متعادل
- پارکینسون
- کوریا (اختلال حرکتی غیرارادی)
- اختلال شناختی
- نوروپاتی محیطی
- تشنج
انواع آتاکسی های مغزی نخاعی کدامند
- ساکاد (saccades): حرکت سریع چشم (پرش)
| نوع آتاکسی | شروع متوسط (محدوده بر حسب سال) | آنچه بیمار تجربه می کند | چالش ها و مسائل مربوط به DNA |
| نوع 1 (SCA1 (ATXN1)) | بین 10 تا 60 سالگی | ساکادهای* هایپرمتریک، ساکادهای آهسته، نورون حرکتی فوقانی (توجه: ساکاد (saccades) مربوط به حرکت چشم است) | تکرار توالی CAG در بازوی کوتاه کروموزوم شماره 6 |
| نوع 2 (SCA2 (ATXN2)) | بین 10 تا 60 سالگی | ساکادهای سرعت کم شده آرفلکسی (عدم رفلکس عصبی) | تکرار توالی CAG در بازوی بلند کروموزوم شماره 12 |
| نوع 3 (SCA3 (ATXN3)) | بین 10 تا 70 سال | بیماری ماچادو جوزف (MJD) نیز نامیده می شود. نیستاگموس برانگیخته ی چشم (حرکت سریع، غیرارادی و نوسانی کره چشم)نورون حرکتی فوقانیساکادهای کند | تکرار توالی CAG در بازوی بلند کروموزوم شماره 14 |
| SCA4 | 19 تا 72 سال | آرفلکسی (عدم رفلکس عصبی) | جهش در بازوی بلند کروموزوم شماره 16 |
چگونه می توان فهمید که به آتاکسی نخاعی مخچه ای مبتلا هستیم یا نه؟
یک پزشک ممکن است آتاکسی مغزی نخاعی را بر اساس موارد زیر تشخیص دهد:
- سابقه خانوادگی این بیماری
- سابقه پزشکی شخص
- معاینه بدنی
- مشاهده علائم مرتبط با SCA
- آزمایش ژنتیک برای افرادی که فکر می کنند ممکن است حامل جهش ژنتیکی SCA باشند، در دسترس است. این آزمایش می تواند به آن ها در تصمیم گیری در مورد تنظیم خانواده و فرزند آوری کمک کند. متخصصین ژنتیک با همکاری متخصصین زنان و زایمان می توانند جنین ها را قبل از به دنیا آمدن از طریق آزمایش های قبل از تولد (مانند آمنیوسنتز) بررسی کنند.
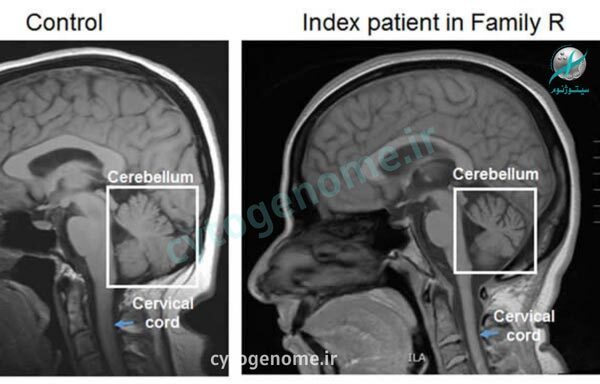
- آزمایش ژنتیک می تواند بسیاری از انواع SCA را تایید کند با این حال، برخی از انواع SCA با یک جهش خاص مرتبط نیستند، بنابراین کارشناسان نمی توانند همه انواع SCA را از این طریق تأیید کنند. در این موارد، پزشکان ممکن است آزمایش ها و عکسبرداری هایی را از مغز برای جستجوی ناهنجاری ها تجویز کنند؛ این آزمایش ها شامل سی تی اسکن و MRI است.
آیا آتاکسی مغزی نخاعی درمان می شود؟
هیچ درمان قطعی برای آتاکسی مغزی نخاعی که در حال حاضر به عنوان یک بیماری پیشرونده و غیرقابل برگشت در نظر گرفته می شود وجود ندارد، اگرچه همه انواع آن به یک اندازه ناتوانی شدید ایجاد نمی کنند. در حالت کلی هدف درمان این بیماری کاهش علائم و بهبود عملکرد بدن فرد بیمار است نه از بین بردن خود بیماری زیرا بسیاری از بیماران مبتلا به اشکال ارثی یا ایدیوپاتیک آتاکسی علاوه بر آتاکسی، علائم دیگری نیز دارند. درمان آتاکسی نخاعی ممکن است شامل موارد زیر باشد:
- وسایل کمکی مانند عصا، واکر یا ویلچر برای کمک به افراد برای حرکت و رفت و آمد کردن.
- فیزیوتراپی برای تقویت عضلات و بهبود راه رفتن و تعادل
- داروهایی برای کاهش لرزش، سفتی و اسپاسم عضلانی.
- به طور معمول فرد مبتلا به این بیماری در نهایت قادر به انجام وظایف روزانه (ADLs) نخواهد بود با این حال، درمانگران توانبخشی می توانند به آن ها کمک کنند تا توانایی خود مراقبتیشان را به حداکثر میزان ممکن برسانند و تا حدودی وخامت بیماری را به تأخیر بیندازند.
محققان هنوز در حال بررسی راه هایی برای کمک به افراد در مدیریت و درمان SCA از جمله RNAi و استفاده از سلول های بنیادی و چندین روش درمانی دیگر هستند.
کلام آخر
آتاکسی مغزی نخاعی بیماری ژنتیکی و ارث پذیری است که موجب طیفی از علائم حرکتی و گفتاری می شود. این اختلال زیر مجموعه هایی دارد که بسته به ژن هایشان شناسایی و درمان می شوند. راهکار تشخیص دقیق انواع آتاکسی ها انجام یک آزمایش ژنتیک هدفمند است. چند نوع از انواع SCA نامشخص باقی مانده اند و نمی توان آن ها را به طور دقیق تشخیص داد، اما در دهه گذشته آزمایش های ژنتیکی امکان شناسایی دقیق ده ها SCA مختلف را فراهم کرده اند و هر سال آزمایش های بیشتر و دقیق تری به این لیست اضافه می شوند. در سال 2008، آزمایش خون ژنتیکی برای بررسی 12 نوع از SCA مانند آتاکسی فریدریش و چندین نوع دیگر اختراع شد. با این حال از آنجایی که همه انواع SCA از نظر ژنتیکی شناسایی نشده اند، برخی از SCA ها هنوز با معاینه عصبی تشخیص داده می شوند که ممکن است شامل معاینه فیزیکی، سابقه خانوادگی، اسکن MRI از مغز و ستون فقرات، و ضربه ستون فقرات باشد.
نویسنده و مترجم: دکتر سارا فرخی مشاور ژنتیک