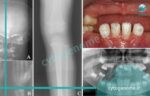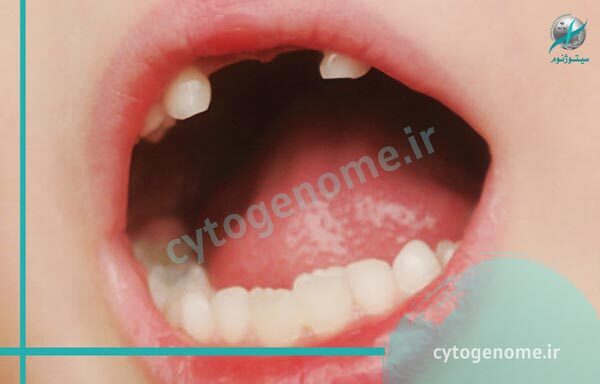
بیماری هیپوفسفاتازی که به آن HPP نیز گفته می شود، استخوان ها را ضعیف و نرم می کند و باعث ناهنجاری های اسکلتی شبیه به بیماری راشیتیسم یا همان نرمی استخوان می شود. نوزادان مبتلا با اندام های کوتاه، قفسه سینه با شکل غیر طبیعی و استخوان های نرم جمجمه متولد می شوند. عوارض بیشتر این بیماری در دوران نوزادی شامل تغذیه نامناسب و عدم افزایش وزن، مشکلات تنفسی و افزایش سطح کلسیم در خون (هیپرکلسمی) است که می تواند منجر به استفراغ مکرر و مشکلات کلیوی شود. این عوارض در برخی موارد تهدید کننده زندگی هستند. در این مقاله قصد داریم در مورد علت هیپوفسفاتازی بحث و گفتگو داشته باشیم، لطفا ما را تا انتها همراهی فرمایید.
چرا به هیپوفسفاتازی مبتلا می شویم؟ علت هیپوفسفاتازی چیست؟
علت هیپوفسفاتازی جهش ژنتیکی در ژن ALPL است و در حال حاضر این ژن تنها ژن شناخته شده ای است که در اثر معیوب شدن، موجب بروز HPP می شود. ژن ALPL نوعی پروتئین به نام آنزیم TNSALP را کُد می کند (آنزیم ها پروتئین های تخصصی هستند که مواد شیمیایی خاصی را در بدن تجزیه می کنند). TNSALP برای رشد و سلامت مناسب استخوان ها و دندان ها ضروری است و در اسکلت، کبد و کلیه ها به وفور یافت می شود. جهش در ژن ALPL منجر به تولید نسخه غیر طبیعی TNSALP می شود و فعالیت این آنزیم را کم می کند به همین دلیل نمی تواند به طور موثر در فرآیند کانی سازی شرکت کند. به این ترتیب TNSALP به نوبه خود منجر به تجمع غیر طبیعی فسفواتانول آمین (PEA)، پیریدوکسال 5’-فسفات (PLP) و پیروفسفات معدنی (PPi) می شود. محققان بر این باورند که تجمع ترکیب پیروفسفات معدنی (PPi)، زمینه ساز اختلال در کانی سازی استخوان ها و دندان ها در افراد مبتلا به هیپوفسفاتازی می شود. جهش های ژن ALPL که تقریباً به طور کامل فعالیت TNSALP را از بین می برد، معمولاً منجر به اشکال شدیدتر هیپوفسفاتازی می شود. سایر جهش ها که فعالیت آنزیم را کاهش می دهند اما آن را بطور کامل از بین نمی برند، اغلب باعث بروز شکل های خفیف تر این بیماری می شوند. به طور کلی، کاهش فعالیت آنزیم TNSALP (که در اثر موتاسیون در ALPL به وجود می آید) با شدت علائم HPP ارتباط دارد (زیرا فعالیت کم آنزیم باعث شدیدتر شدن بیماری می شود).
فراوانی هیپوفسفاتازی چقدر است؟
HPP هم مردان و هم زنان را به میزان مساوی تحت تأثیر قرار می دهد زیرا علت هیپوفسفاتازی جهش در ژن اتوزوم (غیر جنسی) است. تخمین زده شده که در کشورهایی همچون کانادا، HPP شدید در حدود 1 در 100000 تولد زنده رخ می دهد. موارد خفیف تر از این بیماری ممکن است تشخیص داده نشده یا اشتباه تشخیص داده شوند. HPP با بیشترین فراوانی در جمعیت منونیت در کانادا رخ می دهد، در ژاپن نسبتاً شایع است و به نظر می رسد در افراد با اصل و نسب سیاه پوست نادر باشد. به طور کلی هیپوفسفاتازی در سراسر جهان در افراد با نژادهای مختلف گزارش شده است اما خب به نظر می رسد که این اختلال در جمعیت سفیدپوست شایع تر است.
آیا هیپوفسفاتازی ارثی است؟
بله! علت هیپوفسفاتازی بروز یک جهش ژنتیکی در ژنی به نام ALPL است که قابل انتقال به نسول آینده ی فرد مبتلا می باشد. توارث این بیماری می تواند به صورت اتوزومال مغلوب یا اتوزومال غالب باشد. اشکال شدید هیپوفسفاتازمی که در اوایل زندگی ظاهر می شوند (نوع پری ناتال) با الگوی اتوزومال مغلوب به ارث می رسند. وراثت اتوزوم مغلوب به این معنی است که هر دو نسخه ژن که در هر سلول وجود دارد، دچار تغییر و جهش می شود. والدین یک فرد مبتلا به اختلالات اتوزومال مغلوب، هر کدام یک نسخه از ژن جهش یافته را حمل می کنند، اما خودشان علائم و نشانه های این اختلال را نشان نمی دهند (یعنی مادر و پدر ناقل این بیماری هستند و در هر بارداری احتمال 25% وجود دارد که اختلال را به فرزندانشان منتقل کنند). اشکال خفیف تر هیپوفسفاتازی می تواند دارای الگوی توارث اتوزومال غالب باشد؛ وراثت اتوزوم غالب به این معنی است که یک کپی از ژن جهش یافته در هر سلول برای بروز علائم بیماری کافی است. فرم HPP بزرگسالی و ادنتو هیپوفسفاتازی معمولاً توارث اتوزومال غالب دارند و به ندرت پیش می آید که اتوزومال مغلوب باشند. در این توارث، خطر انتقال ژن بیماری زا از والدین مبتلا به فرزندان در هر بارداری 50 درصد خواهد بود.
بیماری هایی که ممکن است با هیپوفسفاتازمی اشتباه گرفته شوند
گذشته از علت هیپوفسفاتازی سایر اختلالات می توانند با این بیماری اشتباه گرفته شوند که در ادامه با آن ها به طور مختصر آشنا خواهید شد:
- بیماری استخوان شکننده: که استئوژنز ایمپرفکتا یا به اختصار OI (Osteogenesis imperfecta) نامیده می شود، گروهی از اختلالات نادر ژنتیکی است که در مجموع شایع تر از HPP می باشد. فرد مبتلا به OI دارای استخوان هایی است که به راحتی در اثر پوکی استخوان شکسته می شوند؛ علائم این بیماری ممکن است خفیف یا شدید باشد. چهار نوع اصلی از بیماری OI شناسایی شده است که نوع I شایع ترین و خفیف ترین شکل و نوع II شدیدترین آن است. اکثر اشکال OI ها به عنوان یک صفت اتوزومال غالب به ارث می رسند.
- راشیتیسم ناشی از کمبود ویتامین D: اختلالی که اغلب در دوران نوزادی یا اوایل کودکی (بعد از 6 ماهگی) آشکار می شود و ناشی از تغذیه نامناسب، عدم قرار گرفتن در معرض نور خورشید، یا سندرمهای سوء جذب است که در آن روده ها مواد مغذی از جمله ویتامین D اضافه شده به برخی غذاها را به اندازه کافی جذب نمی کنند. ویتامین D برای جذب کلسیم و فسفر از روده مورد نیاز است تا سپس به اسکلت رسوب کند. بنابراین، ویتامین D کافی برای رشد مناسب استخوان ضروری است. راشیتیسم ناشی از کمبود ویتامین D باعث نرم شدن استخوان، رشد کند و ضعف عضلانی می شود. این اختلال شایع تر از HPP است و در برخی مناطق محروم در سرتاسر جهان بسیار شایع تر می باشد.
- هیپوفسفاتمی: املای هیپوفسفاتمی شبیه هیپوفسفاتازی است، اما هیپوفسفاتمی به معنای کاهش سطوح فسفات خون می باشد. هیپوفسفاتمی وابسته به X (XLH) شایع ترین شکل ارثی راشیتیسم استئومالاسی است که با ناتوانی کلیه ها در فعال کردن ویتامین D و جذب فسفات از ادرار و جریان خون مشخص می شود؛ همچنین فسفات موجود در غذا ممکن است به خوبی از روده ها جذب نشود. کمبود فسفات خون (هیپوفسفاتمی) منجر به راشیتیسم یا استئومالاسی می شود و رشد فرد مبتلا دچار اختلال شده و اغلب منجر به کوتاهی قد می گردد.
طیف گسترده ای از اختلالات متابولیک استخوان و دیسپلازی های اسکلتی علائمی مشابه HPP دارند که برای تفکیک آن ها از هم لازم است به متخصصین مرتبط برای انجام معاینه، آزمایشات (مانند آزمایش ژنتیک)، نمونه برداری و تصویربرداری پزشکی مراجعه داشت.
نویسنده و مترجم: دکتر سارا فرخی مشاور ژنتیک