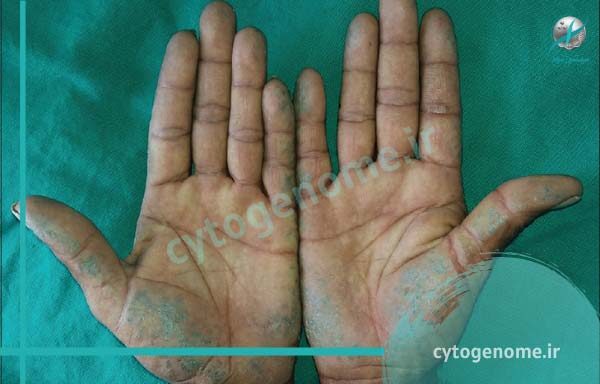
آلکاپتونوری (Alkaptonuria) یا بیماری ادرار سیاه یک اختلال ارثی بسیار نادر است که از تجزیه کامل دو واحد پروتئینی (اسیدهای آمینه) به نام های تیروزین (tyrosine) و فنیل آلانین (phenylalanine) توسط بدن جلوگیری می کند و منجر به تجمع یک ماده شیمیایی به نام اسید هموژنتیسیک (homogentisic acid) در بدن می شود. تجمع این ماده شیمیایی می تواند ادرار و قسمت هایی از بدن را به رنگ تیره تبدیل کند و در طول زمان منجر به بروز طیف وسیعی از مشکلات سلامتی و عوارض جانبی شود. Ochronosis یا تجمع رنگدانه های تیره در بافت های همبند مانند غضروف و پوست نیز مشخصه این اختلال است و این رنگدانه آبی مایل به سیاه معمولاً بعد از 30 سالگی ظاهر می شود. افراد مبتلا به آلکاپتونوریا معمولاً در اوایل بزرگسالی به آرتریت به ویژه در ستون فقرات و مفاصل بزرگ مبتلا می شوند. از دیگر ویژگی های این بیماری می توان به مشکلات قلبی، سنگ کلیه و سنگ پروستات اشاره کرد. تا انتها با سیتوژنوم همراه باشید تا اطلاعات کاملی در رابطه با این بیماری متابولیک کسب کنید.
بیماری آلکاپتونوریا چیست؟
آلکاپتونوری که به آن اختصارا AKU نیز گفته می شود، یک اختلال متابولیک ژنتیکی نادر است و زمانی اتفاق می افتد که بدن انسان نتواند به اندازه کافی آنزیمی به نام دی اکسیژناز هموژنتیزیک (HGD) تولید کند؛ افراد مبتلا فاقد مقدار عملکردی کافی از این آنزیم برای تجزیه هموژنتیزیک اسید هستند که در نهایت با تجمع اسید هموژنتیزیک در بدنشان مواجه می شوند. تجمع اسید هموژنتیسیک باعث تغییر رنگ و شکنندگی استخوان ها و غضروف افراد مبتلا می شود و این امر معمولاً منجر به آرتروز به ویژه در ستون فقرات و مفاصل بزرگ می گردد. افراد مبتلا به آلکاپتونوریا ادراری دارند که وقتی در معرض هوا قرار می گیرد، قهوه ای تیره یا سیاه می شود. البته این تغییر رنگ ممکن است تا چند ساعت پس از ادرار رخ ندهد و اغلب مورد توجه قرار نمی گیرد.
علت AKU چیست؟
آلکاپتونوری در اثر جهش در ژن هموژنتیز 1،2-دیاکسیژناز (HGD) ایجاد می شود. این ژن حاوی دستورالعمل هایی برای ایجاد (رمزگذاری) آنزیمی است که هموژنت 1،2-دیاکسیژناز نام دارد و برای تجزیه اسید هموژنتیزیک ضروری است. جهشهای ژن HGD منجر به کاهش مقدار آنزیم HGD می شود که به نوبه خود منجر به افزایش هموجنتیسیک اسید می گردد. اگرچه این محصول به سرعت توسط کلیه ها از بدن پاک می شود، اما به آرامی در بافت های مختلف بدن به ویژه بافت همبند مانند غضروف تجمع می یابد. با گذشت زمان (به ندرت قبل از بزرگسالی)، در نهایت رنگ بافت آسیب دیده را به آبی یا سیاه تغییر می دهد. تجمع طولانی مدت و مزمن اسید هموژنتیزیک در نهایت باعث ضعیف شدن و آسیب به بافت آسیب دیده می شود و منجر به بسیاری از علائم مشخصه آلکاپتونوری می گردد.
علائم بیماری آلکاپتونوری چیست؟
علائم بیماری آلکاپتونوری از بدو تولد وجود دارد با این حال، علائم بیشتر و چشمگیرتر معمولاً تا بزرگسالی ظاهر نمی شوند. این علائم معمولاً به آرامی پیشرونده هستند و با افزایش سن شدت می یابند. از جمله علائم شایع این بیماری می توان به موارد زیر اشاره کرد:
- ادرار افراد مبتلا به آلکاپتونوریا ممکن است به طور غیرطبیعی تیره باشد یا ممکن است با قرار گرفتن طولانی مدت در مجاورت هوا سیاه شود، با این حال از آنجایی که این تغییر اغلب چندین ساعت طول می کشد، اغلب مورد توجه قرار نمی گیرد. در دوران شیرخوارگی، پوشک ممکن است سیاه رنگ شود (در اثر قرار گرفتن در معرض ادرار در هوا)، اگرچه این مورد اغلب نادیده گرفته می شود.
- در طی سالیان متمادی، اسید هموژنتیزیک به آرامی در بافت های سراسر بدن تجمع می یابد و تقریباً در هر ناحیه ای از بدن، از جمله غضروف، تاندون ها، استخوان ها، ناخن ها، گوش ها و قلب می تواند جمع شود. این تجمع، بافت ها را تیره می کند و باعث ایجاد طیف وسیعی از مشکلات می شود.
- مشکلات مفاصل و استخوان ها: هنگامی که یک فرد مبتلا به آلکاپتونوریا به 30 سالگی می رسد، ممکن است مشکلات مفصلی را تجربه کند که به طور معمول، کمردرد و سفتی و به دنبال آن درد زانو، لگن و شانه خواهد بود؛ این ها علائم اولیه آرتروز هستند. در نهایت، غضروف هایش ممکن است شکننده شده و بشکند و منجر به آسیب مفاصل و ستون فقرات شود که ممکن است جراحی تعویض مفصل برای درمانش مورد نیاز باشد. در حالت کلی افراد مبتلا ممکن است دچار ناهنجاری هایی شوند که بر تاندون ها تأثیر می گذارد، از جمله تاندون های آشیل ضخیم شده و التهاب تاندون ها (تاندونیت). تاندون ها و رباط های آسیب دیده ممکن است در معرض پارگی قرار بگیرند.
- عوارض چشم و گوش: بسیاری از افراد مبتلا روی سفیدی چشم خود لکه های قهوه ای یا خاکستری مشاهده خواهند کرد. علامت دیگر در بسیاری از بزرگسالان مبتلا به آلکاپتونوریا ضخیم شدن غضروف گوش است که همچنین ممکن است آبی، خاکستری یا سیاه به نظر برسد که به آن اکرونوز می گویند. جرم گوش مبتلایان نیز ممکن است سیاه یا قهوه ای مایل به قرمز باشد.
- عوارض پوست و ناخن: آلکاپتونوریا می تواند منجر به تغییر رنگ عرق شود که می تواند لباس ها را لکه دار کند و باعث شود که برخی از افراد دارای نواحی آبی یا سیاه روی پوست خود شوند. رنگ ناخن ها نیز ممکن است مایل به آبی یا قهوه ای شود. تغییرات رنگ پوست نواحی در معرض نور خورشید و جایی که غدد عرق یافت می شوند (مانند گونه ها، پیشانی، زیر بغل و ناحیه تناسلی) آشکارتر است.
- مشکلات تنفسی: اگر استخوان ها و ماهیچه های اطراف ریه ها سفت شوند، می توانند از انبساط قفسه سینه جلوگیری کنند و منجر به تنگی نفس یا مشکل در تنفس شوند.
- مشکلات قلبی، کلیه و پروستات: رسوبات اسید هموژنتیزیک در اطراف دریچه های قلب می تواند باعث سفت شدن و شکننده و سیاه شدن آن ها شود؛ رگ های خونی نیز می توانند سفت و ضعیف شوند. این عارضه می تواند منجر به ایجاد بیماری قلبی شود و ممکن است فرد را نیازمند تعویض دریچه قلب نماید. این رسوب همچنین می تواند منجر به سنگ کلیه، سنگ مثانه و سنگ پروستات شود.
آلکاپتونوری چگونه به ارث می رسد؟
هر سلولی که در بدن انسان های سالم وجود دارد (فارغ از جنسیتشان) شامل 23 جفت کروموزوم (46 عدد) است؛ این کروموزوم ها حامل ژن هایی هستند که افراد از والدین خود به ارث برده اند. یکی از هر جفت کروموزوم از هر یک از والدین به ارث می رسد (به استثنای کروموزوم های جنسی)، به این معنی که دو نسخه از هر ژن در هر سلول وجود دارد. ژن دخیل در ایجاد بیماری آلکاپتونوری ژن HGD نام دارد. این ژن دستورالعمل لازم برای ساخت آنزیمی به نام هموژنتیزات اکسیداز (که برای تجزیه اسید هموژنتیزیک لازم است) را ارائه می دهد. الگوی توارث این بیماری اتوزوم مغلوب است یعنی برای ایجاد آلکاپتونوریا باید دو نسخه از ژن معیوب HGD (یک نسخه از هر والدین) را به ارث ببرید. احتمال بروز این بیماری بسیار اندک است (به همین دلیل است که این وضعیت بسیار نادر می باشد) با این حال در کودکان حاصل از ازدواج های فامیلی شایع تر است. والدین یک فرد مبتلا به آلکاپتونوریا اغلب فقط یک نسخه از ژن معیوب را دارند، به این معنی که هیچ علامت یا نشانه ای از این بیماری را ندارند اما ناقل آن به حساب می آیند. خطر این که دو والد ناقل، هر دو ژن معیوب را به فرزند خود منتقل کنند و در نتیجه فرزندی مبتلا داشته باشند در هر بارداری 25 درصد است. خطر داشتن فرزندی که مانند والدین ناقل است در هر بارداری 50 درصد است. شانس اینکه کودک از هر دو والدین ژن طبیعی دریافت کند و از نظر ژنتیکی برای آن صفت خاص طبیعی باشد 25 درصد است. لازم به ذکر است که این خطر برای مونث و مذکر یکسان می باشد و ربطی به جنسیتشان ندارد.
آلکاپتونوریا چگونه تشخیص داده می شود؟
پزشک عمومی، پزشک متخصص داخلی و یا اورولوژیستی که تحت نظرش هستید ممکن است با شنیدن تغییر رنگ ادرار و مدفوع شما به وجود بیماری آلکاپتونوری مشکوک شود. وی ممکن است برای رسیدن به تشخیص دقیق و سریع تر از تکنیک های زیر کمک بگیرد:
- آزمایش ادراری: مقادیر زیاد هموژنتیزیک اسید در ادرار را می توان با آنالیز کروماتوگرافی گازی – طیف سنجی جرمی تشخیص داد.
- تصویربرداری پزشکی: برای تعیین وجود و میزان بیماری مفصلی و نخاعی یا درگیری دریچه های آئورت یا میترال می توان از تکنیک های مختلف تصویربرداری مانند سی تی اسکن و یا MRI استفاده کرد.
- آزمایش ژنتیک مولکولی: این تست می تواند جهش در ژن HGD را تشخیص دهد با این حال، انجام این آزمایش برای تأیید تشخیص الزامی نیست.
- اکوکاردیوگرافی: در افراد بالای 40 سال، اکوکاردیوگرافی ممکن است برای تشخیص قوی توصیه شود. با اکوکاردیوگرافی، امواج صوتی از قلب منعکس میشوند (پژواک)، که پزشکان را قادر می سازد تا عملکرد و حرکت قلب را بررسی کنند.
نحوه مدیریت و درمان آلکاپتونوریا چگونه است؟
آلکاپتونوری یک بیماری مادام العمر است و در حال حاضر هیچ درمان خاص و قطعی برای آن وجود ندارد. با این حال برای کنترل عوارض و علائمی که ایجاد می کند راهکارهای زیر پیشنهاد می شوند:
- دارو درمانی: افراد مبتلا به آلکاپتونوریا اغلب داروهای ضد التهابی برای درمان درد مفاصل دریافت می کنند و در موارد شدید، ممکن است داروهای قوی تری مانند مواد مخدر برایشان تجویز شود. مدیریت و کنترل درد متناسب با مورد خاص هر فرد تنظیم می شود و نیاز به پیگیری و تنظیم طولانی مدت دارد.
- درمان های حمایتی: برخی از افراد مبتلا به آلکاپتونوریا از فیزیوتراپی و کاردرمانی بهره مند می شوند که می تواند به حفظ قدرت و انعطاف پذیری عضلات و مفاصلشان کمک کند.
- عمل جراحی: برخی از افراد مبتلا به آلکاپتونوریا نیاز به مداخله جراحی دارند به طوریکه تقریباً نیمی از آن ها نیاز به تعویض مفصل ران، زانو یا کتف در سنین 50 تا 60 سالگی خواهند داشت. همچنین به ندرت پیش می آید که این افراد به جراحی ستون فقرات، از جمله فیوژن و یا برداشتن دیسک ها نیاز داشته باشند. جراحی برای تعویض دریچه آئورت یا میترال نیز ممکن است ضروری باشد.
- مصرف ویتامین سی: در کودکان و بزرگسالان، دوزهای بالای ویتامین C نیز برای درمان آلکاپتونوریا استفاده شده است، زیرا مانع از تجمع و رسوب اسید هموژنتیزیک می شود. با این حال مؤسسه ملی سلامت امریکا هشدار می دهد که استفاده طولانی مدت از ویتامین C گاهی اوقات می تواند تولید سنگ کلیه را افزایش دهد و به طور کلی برای درمان طولانی مدت این بیماری بی اثر است.
- داروی نیتیسنون: محققان در حال مطالعه استفاده از دارویی به نام نیتیزینون یا nitisinone (Orfadin®) به عنوان یک درمان بالقوه برای آلکاپتونوری هستند. این دارو که در سال 2001 تاییدیه خود را از سازمان غذا و دارو (FDA) دریافت کرد، برای درمان یک اختلال متابولیک به نام تیروزینمی تایید شده بود. در مطالعات قبلی نشان داده شده بود که نیتیسنون به طور قابل توجهی تجمع اسید هموژنتیزیک را در افراد مبتلا به آلکاپتونوری کاهش می دهد. با این حال، تحقیقات بیشتری برای تعیین اینکه آیا تجویز نیتیزینون برای بیماران جوانتر می تواند از علائم آلکاپتونوری جلوگیری کند یا نه و همچنین برای تعیین ایمنی و اثربخشی طولانی مدت دارو برای افراد مبتلا به AKU ضروری است.
کلام آخر
افراد مبتلا به آلکاپتونوری امید به زندگی طبیعی دارند با این حال، آن ها معمولاً علائم شدیدی مانند درد و از دست دادن توانایی حرکت در مفاصل را تجربه می کنند که به طور قابل توجهی بر کیفیت زندگیشان تأثیر می گذارد. کار و انجام فعالیت های بدنی شدید معمولاً برایشان بسیار دشوار می شود و در نهایت ممکن است برای رفت و آمد به وسایل کمک حرکتی مانند ویلچر نیاز داشته باشند.
نویسنده و مترجم: دکتر سارا فرخی مشاور ژنتیک