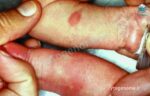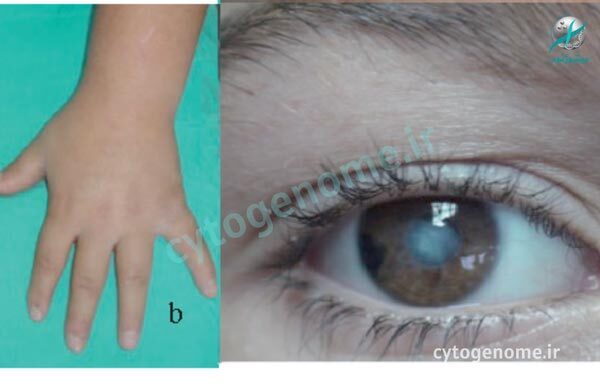
یکی از راهکارهای تشخیص HSAN3 (یا همان دیساآتونومی خانوادگی) معاینه بالینی و بررسی علائم بیمار است. این بیماری که با نام سندروم رایلی دی (Riley Day syndrome) نیز شناخته می شود بر سلول های عصبی (نورون) در سیستم عصبی خودمختار تأثیر می گذارد. بخش خودمختار، بخشی از سیستم عصبی است که عملکردهای غیرارادی مانند تنفس و هضم را کنترل می کند. علائم بیماری HSAN نوع 3 از بدو تولد وجود دارد و شامل مشکل در بلع، کنترل ضعیف فشار خون، دمای بدن و تنفس است. علائم دیگر ممکن است شامل ناتوانی در ایجاد اشک یا احساس درد، استفراغ کردن دوره ای، ذات الریه مکرر و مشکل در راه رفتن باشد؛ به طور کلی با گذشت زمان علائم FD بدتر می شوند و اغلب تهدید کننده زندگی هستند. با این مقاله از سری مقالات ژنتیکی سیتوژنوم همراه باشید تا بیشتر در مورد تشخیص و درمان سندروم رایلی دی (HSAN3 یا دیس آتونومی خانوادگی) اطلاعات کسب کنید.
از کجا بفهمیم که فرزندمان به سندروم رایلی دی مبتلا است یا نه؟
دانستن اینکه فرآیند تشخیص از کجا شروع شود می تواند سخت باشد. همانطور که قبلا نیز اشاره کردیم یکی از راهکارهای تشخیص HSAN3 مشاهده علائم ویژه ای است که نوزدان و خردسالان از خودشان بروز می دهند. در حالت کلی راه هایی که می توان از طریق آن ها به تشخیص HSAN3 رسید عبارت هستند از:
- معاینه بالینی: تشخیص بالینی دیس اتونومی خانوادگی توسط مجموعه ای از علامت های مخصوص امکان پذیر می شود:
- نوزاد پاپیلای قارچی شکل روی زبان خود نداشته باشد
- کاهش رفلکس های عمقی تاندون ها
- فقدان شعله ور شدن آکسون به دنبال تزریق هیستامین داخل جلدی
- مشاهده گریه احساسی بدون سرریز شدن اشک از چشم
- بیوپسی عصبی و عضله ای: بیوپسی (نمونه برداری) عصبی از دست دادن فیبرهای غیر میلین و بیوپسی عضلانی از دست دادن اندام های تاندون گلژی (Golgi tendon organs) را نشان می دهند.
- آزمایش ادرار: دفع کاتکول آمین ها و متابولیت های ادراری کاهش می یابد.
- آزمایش ژنتیک: آزمایش ژنتیک مولکولی یکی از دقیق ترین متدهایی است که می تواند بر تشخیص این بیماری صحه بگذارد. این نوع آزمایش بر روی نمونه کوچکی از خون فرد مشکوک انجام می شود. DNA این فرد با یک کاوشگر (پروب) خاص که از پیش برای جهش های شناخته شده HSAN طراحی شده، بررسی می شود. جالب توجه این است که دقت این نوع تست بالای 99 درصد می باشد.
- آزمایشات قبل از تولد (قبل از زایمان یا پریناتال): دیساآتونومی خانوادگی با الگوی اتوزومال مغلوب به ارث می رسد، به این معنی که دو نسخه از ژن مورد نظر در هر سلول تغییر کرده و معیوب می شود. اگر با انجام آزمایشات ژنتیکی نشان داده شود که هر دو والدین ناقل این بیماری هستند، احتمال ابتلای کودکانشان به این بیماری 25 درصد خواهد بود. برای بارداری هایی که در معرض خطر FD هستند، تشخیص ژنتیکی قبل از لانه گزینی (PGD) یا تشخیص قبل از تولد با آمنیوسنتز (که در هفته های 15-17 انجام می شود) یا نمونه برداری از پرزهای کوریونی (یا همان CVS در هفته های 10-14 بارداری) امکان پذیر است.
نکات بالینی برای تشخیص هرچه بهتر بیماری دیس اتونومی خانوادگی
خوب است برای تشخیص HSAN3 این نکات را نیز در نظر بگیرید:
- در اوایل دوران نوزادی، بلع ضعیف همراه با ناهماهنگی حلق و آسپیراسیون وجود دارد. مکیدن ضعیف و مشکلات بلع، مشکل اصلی است که منجر به شکست قابل توجهی در رشد می شود.
- عرق کردن هنگام غذا خوردن
- راه رفتن تاخیری و آتاکسیک نیز از این بیماری گزارش شده است. آتاکسی ثانویه به از دست دادن بازخورد توسط دوک عضلانی است.
- به تدریج، درد و حس حرارتی از بین می رود.
- هنگامی که کودکان 2 تا 6 ساله هستند، زخم قرنیه بدون درد و صدمات تروماتیک (بدون احساس درد) به طور مکرر رخ خواهد داد.
- رفلکس های کششی تاندون وجود ندارد
- تغییر شکل اسکلتی و اسکولیوز اغلب وجود دارد
- تشنج عمومی را می توان در 40 درصد از این بیماران مشاهده کرد.
آیا سندروم رایلی دی ارث پذیر است؟
یکی از راهکارهای تشخیص HSAN3 این است که به مشاور ژنتیک جهت رسم شجره نامه مراجعه کنید. همانطور که احتمالا می دانید، الگوی توارث این بیماری اتوزومال مغلوب با نفوذ کامل است. بنابراین ما این بیماری را بیشتر در کودکانی می بینیم که والدینشان با یکدیگر نسبت فامیلی دارند. احتمال ابتلای این کودکان به این بیماری (در صورت ناقل بودن والدین) 25% می باشد. دو جهش در ژنی به نام IKBKAP می تواند باعث سندروم رایلی دی (HSAN3) شود:
- جهش در اینترون 20 (99٪ موارد) که در آن محصول ژن IKAP در مغز بیان نمی شود (اما در لنفوبلاست ها وجود دارد)
- جهش بی معنی (missense mutation) در اگزون 19 که باعث اختلال در محل فسفوریلاسیون IKAP می شود که فعالیت آن را کاهش می دهد.
آیا درمان سندروم رایلی دی امکانپذیر است؟
در حال حاضر هیچ راهکار قطعی برای درمان سندروم رایلی دی وجود ندارد. تنها دو مرکز درمانی در خارج از ایران وجود دارد که در حال برنامه ریزی برای درمان این بیماری هستند. شاید با خواندن این جمله ناامید شده باشید اما باید بدانید اگرچه ژن ایجاد کننده این سندروم شناسایی شده است و به نظر می رسد بیان خاصی از بافت دارد، اما هیچ درمان قطعی فعلا برای آن در هیچ جای دنیا وجود ندارد. از آنجایی که علائم بیماری دیس اتونومی خانوادگی در بیماران مختلف، ثابت و یکسان نیست (یعنی نوع و شدت علائم در بین بیماران و حتی در سنین مختلف در همان بیماران متفاوت است) بنابراین مجبوریم درمان های پیشگیرانه، علامتی و حمایتی برایشان در نظر بگیریم. این راهکارهای درمانی عبارت اند از:
- همیشه یک مسئله اصلی در مورد بیماران HSAN3، پنومونی آسپیراسیون بوده است. بنابراین استفاده از فوندوپلیکاسیون (با جلوگیری از نارسایی) و لولههای گاستروستومی (برای ارائه تغذیه غیر خوراکی) می تواند تعداد دفعات بستری شدن در بیمارستان را کاهش بدهد به نوعی درمان سندروم رایلی دی محسوب می شود.
- پزشکان معالج می توانند از داروهای مخصوص برای کنترل استفراغ، خشکی چشم و فشار خون غیرطبیعی استفاده کنند (مانند دیازپام داخل وریدی یا رکتوم، یا کلرال هیدرات رکتوم)
- استراتژی تغذیه ای مناسب برای حفظ تغذیه کافی
- فیزیوتراپی روزانه قفسه سینه (نبولیزاسیون، گشادکننده برونش و درناژ وضعیتی) برای بیماری مزمن ریوی و رفع مشکلات استخوان و مفصلی با ارتوپدی
- جلوگیری از آسیب های تصادفی
- پیشگیری از افت فشار خون ارتواستاتیک (با انجام هیدراتاسیون، ورزش پا، وعده های غذایی کوچک مکرر، رژیم غذایی پر نمک، و مصرف داروها (مثلاً با فلودروکورتیزون)،
- جبران فشار خون ناپایدار
والدین و بیماران باید در مورد مراقبت روزانه از چشم و علائم اولیه مشکلات قرنیه و همچنین کوتر نقطه ای مطلع شوند. اطلاع رسانی به بیماران و مراقبین منجر به کاهش اسکار قرنیه و نیاز به اقدامات جراحی تهاجمی تر مانند تارسرافی، فلپ ملتحمه و پیوند قرنیه شده است.
خلاصه
دیساآتونومی خانوادگی (Familial dysautonomia یا FD) که همچنین با نام های سندروم رایلی دی (Riley Day syndrome) و نوروپاتی ارثی حسی و اتونومیک نوع 3 (HSAN3) شناخته می شود، یک اختلال ژنتیکی نادر، پیش رونده در سیستم عصبی خودمختار است که بر رشد و بقای حسی، سمپاتیک و برخی اعمال پاراسمپاتیک تأثیر می گذارد. اختلال در نورون ها (سلول های عصبی) در سیستم عصبی منجر به علائم متغیری از جمله عدم حساسیت به درد، ناتوانی در تولید اشک، رشد ضعیف و فشار خون ناپایدار (فشار خون اپیزودیک و افت فشار خون وضعیتی) می شود. افراد مبتلا به HSAN نوع 3 دچار بحران های استفراغ، ذات الریه، مشکلات گفتاری و حرکتی، مشکل در بلع، درک نامناسب گرما، درد و طعم و همچنین فشار خون ناپایدار و اختلال حرکتی دستگاه گوارش هستند. تشخیص HSAN3 بر اساس ویژگی های بالینی است: کاهش اشک، عدم حساسیت به درد، کنترل ضعیف دما، از بین رفتن رفلکس های تاندون عمیق، افت فشار خون وضعیتی، حملات استفراغ، هماهنگی حرکتی ضعیف و عقب ماندگی ذهنی. درمان آن نیز علامتی می باشد؛ لازم به ذکر است که بسیاری از کودکان در سال های اول زندگی معمولاً در نتیجه پنومونی آسپیراسیون مکرر می میرند.
نویسنده و مترجم: دکتر سارا فرخی مشاور ژنتیک