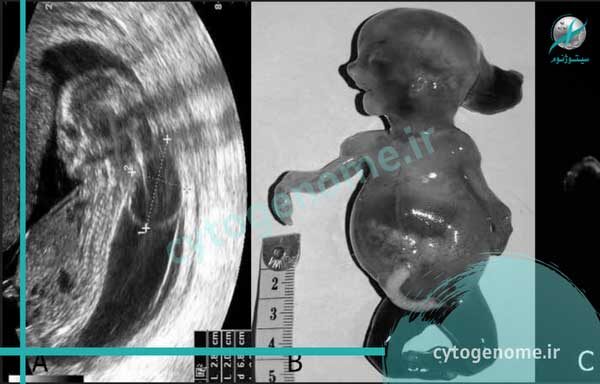
سندروم مکل یک بیماری ژنتیکی بسیار شدید است که با کیست های متعدد روی کلیه ها، بیرون زدگی بخشی از مغز از طریق سوراخی در جمجمه (آنسفالوسل پس سری) و وجود انگشت اضافه روی دست یا پا (پلی داکتیلی) مشخص می شود. کودکان مبتلا همچنین ممکن است ناهنجاری هایی داشته باشند که بر سر و صورت، کبد، ریه ها، اندام تناسلی و دستگاه ادراریشان تأثیر بگذارد. این سندروم در اثر تغییرات ژنتیکی در یکی از ژن های شناسایی شده ایجاد می شود و به صورت اتوزومال مغلوب به ارث می رسد. با این مقاله از سیتوژنوم همراه باشید تا اطلاعات بیشتری در مورد علائم سندروم مکل و توارث آن کسب نمایید.
سندروم مکل چیست؟
سندروم مکل (Meckel syndrome) که با نام سندروم مِکِل گروبر (Meckel Gruber syndrome) نیز شناخته می شود، یک ناهنجاری شدید و پیچیده ژنتیکی است که دختران و پسران را درگیر می کند و بر چندین سیستم اندامی بدن تأثیر می گذارد. نام این سندروم از اولین پزشکی که آن را شناسایی و توصیف کرد گرفته شده است. اولین گزارش سندرم مکل توسط جان فردریک مکل (Johann Friedrich Meckel) در سال 1822 منتشر شد و در سال 1934 پزشکی به نام گروبر گزارش هایی را در مورد افراد مبتلا به سندرم مکل منتشر کرد و نام این اختلال را dysencephalia splanchnocystica گذاشت. آن ها اتفاق نظر داشتند که سه علامت کلاسیک در افراد مبتلا به این بیماری رایج است و سلامتی آن ها را به شدت تحت تاثیر قرار می دهد.
مترادف های سندروم مکل
در زبان ژنتیکی و پزشکی، سندروم مکل را با نام های زیر نیز ممکن است معرفی کنند:
- دیسنسفالی اسپلانکنو سیستیکا (Dysencephalia splanchnocystica)
- سندرم گروبر (Gruber syndrome)
- سندرم مکل گروبر (Meckel-Gruber syndrome)
- MES
- MKS
طول عمر افراد مبتلا به سندروم مکل چقدر است؟
به دلیل مشکلات جدی سلامتی، نوزادانی که با سندروم مکل متولد می شوند بیش از چند روز یا چند هفته زنده نمی مانند و اکثر نوزادان مبتلا به نارسایی کلیه یا مشکلات تنفسی می میرند. گاهی اوقات وقتی جنین مبتلا به سندرم مِکِل در دوران بارداری تشخیص داده می شود، والدین تصمیم می گیرند که حاملگی را به طور قانونی خاتمه دهند تا از به دنیا آمدن یک کودک ناهنجار و تحمل بار عاطفی شدید جلوگیری نمایند.
توارث و ژن های موثر در سندروم مکل
سندروم مکل به عنوان یک بیماری با الگوی توارث اتوزومال مغلوب به ارث می رسد. طبق آخرین تحقیقات مشخص شده که این بیماری از طریق سیزده ژن به ارث می رسد که عبارت هستند از:
- B9D1
- B9D2
- CC2D2A
- CEP290
- MKS1 (رایج ترین ژن)
- RPGRIP1L
- TCTN2
- TCTN3
- TMEM67
- TMEM107
- TMEM216
- TMEM2
علائم و نشانه ها
علائم خاص مرتبط با سندروم مکل از فردی به فرد دیگر بسیار متفاوت است؛ به همین دلیل است که کودکان مبتلا همه علائمی که در زیر توضیح داده شده است را نخواهند داشت. علائمی که تا به الان از این سندروم گزارش شده است عبارت هستند از:
- آنسفالوسل اکسیپیتال: شایع ترین ناهنجاری موجود در سیستم عصبی مرکزی در اثر ابتلا به سندرم مکل، آنسفالوسل اکسیپیتال است. این عارضه وضعیتی است که طی آن نوزاد با شکافی در جمجمه خود متولد می شود (یعنی بخشی از یک یا چند صفحه استخوانی که جمجمه را تشکیل می دهند به هم متصل نمی شوند)؛ به این ترتیب غشاهایی که مغز (مننژها) و بافت مغز را می پوشانند اغلب از این شکاف بیرون زده می شود. انسفالوسل اکسیپیتال ممکن است منجر به تجمع بیش از حد مایع مغزی نخاعی (CSF) در جمجمه شود که باعث فشار بر بافت های مغز (هیدروسفالی) می گردد.
- سایر ناهنجاری های سیستم عصبی مرکزی: دیگر ناهنجاری های موجود در سیستم عصبی مرکزی که ممکن است در نوزادان مبتلا به سندرم مکل دیده شود شامل فقدان بخش بزرگی از مغز، جمجمه و پوست سر (آنسفالی)، فقدان قسمت میانی مغز خلفی (مخچه vermis agenesisi) و یک بیماری به نام میکروسفالی است (که در آن دور سر نوزاد کوچکتر از حد انتظار برای سن و جنسیت اوست).
- ناهنجاری های ظاهری: نوزادان مبتلا ممکن است دارای ویژگی های متمایز صورت از جمله کوچک بودن غیرطبیعی فک (micrognathia)، گوشهای بزرگ و نامتقارن و بدشکل؛ شکاف کام و شکاف لب؛ پیشانی شیب دار، گردن کوتاه داشته باشند.
- علائم چشمی: کودکان مبتلا ممکن است ناهنجاری های چشمی از جمله چشم های کوچک غیرطبیعی (میکروفتالمی) و توسعه نیافتگی اعصاب چشم (هیپوپلازی عصب بینایی یا کولوبوما) داشته باشند.
- کلیه پلی کیستیک: وجود کیست های متعدد روی کلیه ها (دیسپلازی کلیه پلی کیستیک) شایع ترین علامت مرتبط با سندرم مکل است. این وضعیت در طی سونوگرافی مشخص می شود و پزشک خواهد دید که کلیه بافت طبیعی دارد اما با کیسه های پر از مایع یا کیست هایی با اندازه های مختلف پر شده است که با پیشرفت بیماری بزرگتر می شوند (10 تا 20 برابر بیشتر از حد طبیعی). یافته های مرتبط با کلیه های کیستیک شامل از دست دادن عملکرد کلیه است که منجر به نارسایی کلیه در مرحله نهایی می شود. عملکرد نامناسب کلیه همچنین منجر به کاهش مقدار مایع آمنیوتیک اطراف جنین در حال رشد (الیگوهیدرآمنیوس) می شود.
- پلی داکتیلی: افراد مبتلا همچنین ممکن است انگشتان اضافی بر روی دست و پا خود داشته باشند. اغلب این انگشتان اضافی در سمت انگشت کوچک دست (پلی داکتیلی پساکسیال) دیده می شوند.
- ناهنجاری های اسکلتی: ناهنجاری های اسکلتی عبارت اند از: خم شدن استخوان های بلند بازوها و پاها، انحنای انگشتان پنجم (کلینوداکتیلی)، در هم بافته شدن انگشتان دست و پا (سینداکتیلی) و پای چاقویی که در آن پا به صورت داخلی می چرخد (talipes equinovarus).
- ناهنجاری های تناسلی: در برخی افراد مبتلا، ناهنجاری هایی در دستگاه تناسلی ادراری ممکن است وجود داشته باشد که از جمله آن ها می توان به عدم نزول یک یا هر دو بیضه به داخل کیسه بیضه (کریپتورکیدیسم)، مثانه توسعه نیافته (هیپوپلاستیک) و رشد ناقص دستگاه تناسلی اشاره کرد. ناهنجاری های تناسلی شامل دستگاه تناسلی خارجی یا داخلی در هر دو جنس مرد یا زن است.
- ناهنجاری اندام های داخلی: برخی از نوزادان مبتلا ممکن است ناهنجاری هایی داشته باشند که سایر اندام های بدن از جمله کبد، ریه ها یا قلب را تحت تاثیر قرار دهند. کبد می تواند بافت فیبری بیش از حد (فیبروز)، گشاد شدن (اتساع) و افزایش تعداد (تکثیر) مجاری کوچکی را که صفرا را از کبد به روده های کوچک (مجاری صفراوی) می برد، نشان دهد. ریه ها نیز ممکن است توسعه نیافته باشند (هیپوپلاستیک)؛ همچنین ساختاری که ورودی حنجره را هنگام بلع می پوشاند ممکن است شکاف داشته باشد (شکاف اپی گلوت). طحال ممکن است از بین رفته باشد (آسپلینیا)، یا به صورت چندین طحال کوچک دیده شود تا یک طحال کامل و سالم (پلی اسپنی).
- ناهنجاری های قلبی: ناهنجاری های قلبی ممکن است شامل نقایص سپتوم دهلیزی و بطنی (ASDs و VSDs) و مجرای شریانی باز یا سایر ناهنجاری های پیچیده تر باشد. ASD ها با یک باز شدن غیر طبیعی در پارتیشن فیبری (سپتوم) که دو حفره فوقانی (دهلیز) قلب را از هم جدا می کند، مشخص می شوند. VSD ها با یک باز شدن غیر طبیعی در سپتوم مشخص می شوند که دو حفره پایینی قلب (بطن) را تقسیم می کند. اندازه، محل و ماهیت نقص سپتوم و هر گونه ناهنجاری مرتبط با آن، شدت علائم را تعیین می کند. باز بودن یا شل بودن مجرای شریانی وضعیتی است که در آن مجرای بین رگ خونی که به ریه ها (شریان ریوی) و شریان اصلی بدن (آئورت) منتهی می شود، پس از تولد بسته نمی شود.
نویسنده و مترجم: دکتر سارا فرخی مشاور ژنتیک

