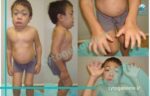
بیماری موکوپلی ساکاریدوز نوع یک (Mucopolysaccharidosistype I) از جمله بیماری های ژنتیکی ارث پذیری است که موجب ایجاد طیف وسیعی از علائم در نقاط مختلف بدن می شود. این اختلال در اثر نقص در ژنی به نام IDUA ایجاد می شود که مسئول تجزیه کردن قندهای بزرگی به نام گلیکوز آمینو گلیکان ها (یا به اختصار GAGs) هستند. هنگامی که این ژن به درستی کار نکند، آنزیمی به نام IDUA نیز تولید نمی شود یا کمتر از حد طبیعی تولید می شود که منجر به افزایش اندازه لیزوزوم ها (نوعی اندامک سلولی) و اندام های بدن (ارگانومگالی) می شود. در این مقاله خواهید خواند که نحوه توارث، فراوانی، تشخیص و درمان موکوپلی ساکاریدوز نوع 1 چگونه خواهد بود.
فراوانی موکوپلی ساکاریدوز نوع 1 چقدر است؟
قبل از آنکه در مورد درمان موکوپلی ساکاریدوز نوع 1 توضیح بدهیم باید بدانید که این بیماری نسبتا نادر است و در برخی از کشورهای توسعه یافته کم دیده می شود. MPS I نوع شدید، تقریباً از هر 100000 نوزاد یک نفر را درگیر می کند و نوع ضعیف آن (که کمتر شایع است) در 1 نفر از هر 500000 تولد دیده می شود. به این ترتیب محققان اعلام کرده اند که سندروم هارلر 57 درصد موارد، سندرم هارلر- شی 23 درصد و سندروم شی 20 درصد موارد ابتلای بیماری موکوپلی ساکاریدوز را تشکیل می دهند.
آیا موکوپلی ساکاریدوز ارث پذیر است؟
بله! بیماری MPS نوع 1 بیماری ژنتیکی است که با الگوی توارث اتوزومال مغلوب (autosomal recessive) به ارث می رسد. به این معنی که هر دو نسخه موجود از ژن IDUA (که در هر سلول از بدن وجود دارند) دارای جهش هستند و به عبارتی، معیوب یا غیر طبیعی می باشند. والدین یک فرد مبتلا به بیماری که توارث اتوزوم مغلوب دارد، هر کدام یک نسخه از ژن جهش یافته را حمل می کنند (یعنی ناقل هستند)، اما معمولاً علائم و نشانه های این بیماری را نشان نمی دهند. در مورد بیماری موکوپلی این احتمال وجود ندارد که مبتلایان بزرگ شوند و به سن باروری برسند؛ بنابراین والدین بیمار (کسانی که علائم نشان دهند) وجود نخواهد داشت.
تشخیص موکوپلی ساکاریدوز نوع 1 ابتدای تولد
تشخیص زودهنگام این اختلال دشوار است زیرا اولین علائم بالینی که در نوزادان دیده می شوند خاص نیستند (فتق، عفونت های تنفسی و غیره) و آن ها را می توان به بسیاری از اختلالات رایج دیگر در ابتدای تولد نسبت داد با این حال اما بسیار مهم است که امکان درمان موکوپلی ساکاریدوز نوع 1 به سرعت فراهم شود. به هرحال تشخیص MPS نوع I بر اساس شناسایی علائم بروز داده شده، شرح حال دقیق از بیمار و خانواده، ارزیابی بالینی کامل و انجام انواع آزمایشات تخصصی است. گاهی اوقات ممکن است پزشک علائم اولیه مشکوکی را در نوزادان ببیند که به وجود این بیماری مشکوک شود. MPS 1 معمولاً در افرادی که حداقل یکی از دو یافته زیر را دارند، در ابتدای تولد تشخیص داده می شود:
- فعالیت کم آنزیم آلفا ال- ایدورونیداز
- شناسایی جهش ژنی از طریق توالی یابی ژن IDUA
- افزایش سطح GAG در ادرار و یا خون نوزاد
تشخیص دقیق موکوپلی ساکاریدوز نوع 1 چگونه است؟
به منظور انتخاب یک راهکار مناسب برای شروع درمان موکوپلی ساکاریدوز نوع 1 ابتدا باید این بیماری به طور دقیق شناسایی و تایید شود. به منظور رسیدن به قطعیت می توان از متدهای زیر برای تشخیص استفاده نمود:
- تست و کار بالینی
بررسی اختصاصی فاکتورهای آنزیمی و بیوشیمیایی در ادرار افراد مبتلا به MPS I بسیار مهم است. ادرار این افراد به طور معمول سطوح بالای گلیکوز امینو گلیکان ها (موکوپلی ساکاریدها)، به ویژه هپاران و سولفات درماتان را نشان می دهد. درست است که این فاکتور به معنای تشخیص نهایی MPS I نیست، اما نشان دهنده وجود یک اختلال موکوپلی ساکاریدوز در فرد خواهد بود.
- بررسی فعالیت آنزیم IDUA
تشخیص قطعی MPS I مستلزم انجام آزمایش سلول های خاص مانند گلبول های سفید (لکوسیت ها) یا سلول های بافت همبند (فیبروبلاست ها) است. این آزمایش ها فعالیت کم یا طبیعی آنزیم آلفا ال-ایدورونیداز را نشان می دهند.
آزمایش ژنتیک مولکولی اغلب برای تأیید نهایی تشخیص استفاده می شود. این نوع آزمایش می تواند جهش هایی در ژن IDUA که عامل MPS I شناخته شده و در ایجاد بیماری موکوپلی نقش دارند را شناسایی کند، اما فقط به عنوان یک گزینه تشخیصی تقریبا پر هزینه در آزمایشگاه های تخصصی ژنتیک پزشکی در دسترس است.
- غربالگری نوزادان تازه به دنیا آمده
یکی از مشکلات غربالگری نوزادان برای تشخیص MPS I این است که در برخی موارد پزشکان ممکن است نتوانند تشخیص دهند که آیا نوزاد مبتلا به MPS I به شکل شدید این اختلال مبتلا هستند یا ضعیف. از آنجایی که درمان اشکال مختلف موکوپلی متفاوت است، این امر می تواند منجر به ایجاد چالش هایی در تعیین دوره مناسب برای درمان شود. تحقیقات در حال حاضر برای یافتن روش هایی برای تفکیک نوزادان مبتلا به MPS I شدید از نوزادان با MPS I ضعیف در حال انجام است.
- تشخیص قبل از زایمان (تشخیص پریناتال)
در خانواده هایی که از قبل وجود این جهش تایید شده است، تشخیص قبل از تولد می تواند با سنجش آنزیمی یا ژنتیک مولکولی انجام شود.
- تشخیص های افتراقی
موکوپلی ساکاریدوز نوع VI (یا سندروم Maroteaux Lamy) از بسیاری جهات به موکوپلی ساکاریدوز نوع I شباهت دارد، با این حال بیماران MPS VI، هرگز دچار اختلال فکری نمی شوند. موکوپلی ساکاریدوز نوع II یک بیماری با توارث وابسته به ایکس مغلوب X است که در آن انقباضات شدید مفصلی یک علامت مشخصه در مبتلایان می باشد، همچنین دارای ویژگی های مشترک بسیاری با موکوپلی ساکاریدوز نوع I است.
آیا درمان موکوپلی ساکاریدوز نوع 1 امکانپذیر است؟
هیچ راهکاری معرفی نشده است که به طور کامل موجب درمان موکوپلی ساکاریدوز نوع 1 شود با این حال، درمان های موثری وجود دارد که ثابت شده است پیشرفت بیماری را کند می کنند. به عنوان مثال پیوند سلول های بنیادی خونساز (HSCT) می تواند به برخی از بیماران مبتلا به فرم شدید موکوپلی کمک کند و به طور کلی انجام آن در 1 الی 2 سال اول زندگی توصیه می شود. در حالت کلی سه جزء در درمان MPS I وجود دارد:
- جایگزینی آنزیمی که از دست رفته است
- کاهش علائم خاص بیماری
- ارائه مشاوره ژنتیک به خانواده فرد مبتلا
در ادامه این درمان ها را به تفصیل بررسی می کنیم:
آنزیم های لیزوزومی پروتئین های منحصر به فردی هستند زیرا می توانند توسط سلول ها جذب شده و مورد استفاده قرار گیرند. به این ترتیب یکی از راه های بالقوه برای درمان MPS I، دادن آنزیم آلفا ال-ایدورونیداز به بیماران است که این کار به 2 روش قابل انجام می باشد. یکی از راه ها تزریق آنزیم خالص شده (ERT) است؛ درمان ERT شامل جایگزینی آنزیم از دست رفته آلفا ال-ایدورونیداز، توسط یک متد مهندسی شده ژنتیکی (نوترکیب) است. این کار با تزریق آنزیم به بیمار به صورت هفتگی از طریق داخل وریدی انجام می شود. در سال 2003، سازمان غذا و داروی ایالات متحده امریکا (FDA) لارونیداز (Aldurazyme) را برای درمان اکثر افراد مبتلا به MPS I تایید کرد.
روش دیگر برای جایگزینی آنزیم در بیماران، انجام پیوند سلول های بنیادی خونساز (HSCT) یا پیوند مغز استخوان است. سلول های بنیادی خون ساز سلول های تخصصی هستند که در مغز استخوان (ماده اسفنجی نرم موجود در استخوان های بلند) یافت می شوند. این سلول ها در خون رشد می کنند و در نهایت به یکی از سه نوع اصلی گلبول های خونی (یعنی گلبول های قرمز، گلبول های سفید یا پلاکت ها) تبدیل می شوند. مغز استخوان مورد نیاز برای پیوند به فرد مبتلا از فردی گرفته می شود که MPS I ندارد (معمولا خواهر و یا برادر). با این حال این روش گران است و خطر عوارض جدی از جمله مرگ، بیماری و سایر عوارض طولانی مدت و دیررس را به همراه دارد. HSCT به عنوان استاندارد مراقبت برای MPS I شدید در نظر گرفته می شود، زیرا تنها درمان موجود است که نشان داده منجر به بهبود نتایج دهنی در افراد مبتلا به نوع شدید MPS I می شود. اگرچه درمان با پیوند مغز استخوان و آنزیم می تواند به طور چشمگیری سیر بالینی را بهبود بخشد، اما این اختلال را درمان نمی کند و بنابراین، بسیاری از افراد مبتلا همچنان با افزایش سن با چالش ها و مسائل قابل توجهی روبرو هستند. به طور کلی، هرچه درمان زودتر شروع شود نتیجه بهتر خواهد بود زیرا سن کودک اغلب بر نتیجه عمل تأثیر می گذارد.
- کاهش علائم خاص بیماری:
یک جزء مهم از درمان موکوپلی ساکاریدوز نوع 1، کنترل یا درمان علائم خاصی است که هر فرد از خود بروز می دهد. این نوع درمان مستلزم تلاش هماهنگ تیمی از متخصصانی شامل متخصص اطفال، قلب، اعصاب، چشم پزشک، ارتوپد، روماتولوژیست، فیزیوتراپیست، کار درمانگر، گوش و حلق و بینی و ریه می باشد.
- مشاوره ژنتیک
مشاوره ژنتیک به افراد مبتلا و خانواده آن ها توصیه می شود. یک متخصص ژنتیک می تواند با رسم شجره نامه و انجام آزمایش ژنتیک باعث شود که دیگر فرزندان مبتلا در خاندان آن ها به دنیا نیاید و به عبارتی ریشه این بیماری در خانواده ها سوزانده شود.
نویسنده و مترجم: دکتر سارا فرخی مشاور ژنتیک